











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Membranous nephropathy is rare in the paediatric age group, with an estimated incidence of 0.1/100 000 per year compared to 1.2/100000 per year in the adult population.1 It describes a specific histopathologic pattern of glomerular lesion, rather than a single disease entity with a common pathophysiology. The term “membranous” refers to an apparent thickening of glomerular capillary walls by optical microscopy due to the subepithelial deposition or in situ formation of immune complexes.2 Recent progress in our understanding of the disease has been made as new target antigens in human podocytes are being identified. This has allowed for more precise nomenclature and classification. Currently, membranous nephropathy is considered primary when a humoral autoimmune response against a normal podocyte antigen is implied in the absence of a secondary cause.
While most cases in the paediatric age group are secondary, adult disease is more frequently primary. The M-type phospholipase A2 receptor (PLA2R) has been identified as the antigen target in most cases of primary membranous nephropathy in adults. Circulating autoantibodies against PLA2R (anti-PLA2R) and co-localization of IgG and PLA2R in renal tissue are now being used to help distinguish primary membranous nephropathy from secondary cases.3 Measuring titres of anti-PLA2R is also in use to monitor the immunological activity during treatment, improving the prognostic tools to tailor immunosuppression.2,4Whereas immunosuppression is often needed to treat primary disease, management of secondary membranous nephropathy has been mainly based on early recognition and treatment of the underlying cause.5,6As the literature on paediatric membranous nephropathy is limited and knowledge has been mainly extrapolated from adult studies, we report a case of an adolescent treated with penicillamine for Wilson’s disease who presented with nephrotic syndrome due to membranous nephropathy.
„
CASE REPORT
We report the case of a 16-year-old girl who presented with nephrotic syndrome fourteen months after penicillamine initiation for treatment of Wilson’s disease. The clinical course of the patient over time is summarized in Fig. 1.
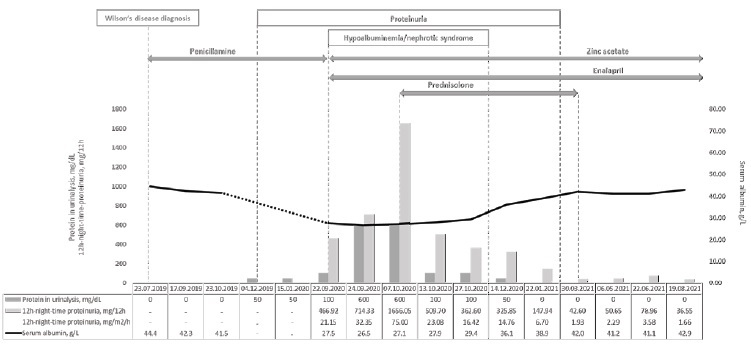
She was the second female child born to a non-consanguineous Caucasian couple. Her family history was positive for dyslipidaemia, but otherwise unremarkable. Wilson’s disease had been diagnosed based on persistently elevated alanine aminotransferase (ALT) levels in the range of 100 to 200 U/L (reference interval: 9 - 24 U/L) and aspartate aminotransferase (AST) levels in the range of 50 to 100 U/L (reference interval: 13 - 26 U/L) since the age of 14, hepatomegaly (18 cm), neuropsychiatric symptoms (anxiety and declining performance at school), Kayser-Fleischer rings, low serum ceruloplasmin <0.02 g/L (reference range: 0.2 - 0.6 g/L), and excessive 24-hour urinary excretion of cooper 95 mcg/24 hour (normal <40 mcg/24h). As the diagnosis of Wilson’s disease was highly likely according to the Ferenci score,7 treatment with penicillamine was initiated. As recommended, pyridoxine supplementation was also started.
At five months after decoppering therapy initiation, the urinalysis started to show isolated proteinuria (50 mg/dL). A decision to keep treatment under close observation was made, considering that the patient was otherwise well and showed a very effective clinical and laboratorial response to Wilson’s disease treatment. The urinalysis continued to show the same level of isolated proteinuria (50 mg/dL) at six and at nine months of follow-up.
At fourteen months after penicillamine initiation, the patient presented to the emergency department with increasing weight (from 68 to 72 kg) and progressively worsening oedema for the previous three weeks. There was no history of recent fever or any other symptoms of intercurrent illness, immunizations or exposure to new drugs or toxics. Her physical exam was positive for bilateral periorbital oedema and bilateral lower extremities pitting oedema. Her blood pressure was 105/60 mmHg, which was normal according to the reference values for age, sex and height (<50th percentile for both systolic and diastolic blood pressure).8 The urinalysis showed aggravated proteinuria (100 mg/dL) without haematuria, and the urinary sediment was unremarkable. The quantitative measurement of proteinuria was obtained through a 12-hour night-time urine collection, which revealed a protein excretion of 466 mg, corresponding to 21 mg/m2/hour (for reference, urinary protein excretion in excess of 4 mg/m2/hour is considered abnormal in children and nephrotic range proteinuria is defined as a urinary protein excretion above 40 mg/m2/hour). The remaining laboratory workup showed hypoalbuminemia (27.1 g/L) and hypercholesterolemia (total cholesterol 381 mg/dL, LDL-cholesterol 242 mg/dL, HDL-cholesterol 99 mg/dL). Serum creatinine was 0.61 mg/dL (normal range for age: 0.59 - 0.88 mg/dL). Accordingly, the estimated glomerular filtration rate was 148 mL/min/1.73 m2.
Penicillamine was withhold and zinc acetate was initiated as maintenance treatment for Wilson’s disease. Sodium-restricted diet, furosemide, enalapril and simvastatin were implemented as part of the supportive management.
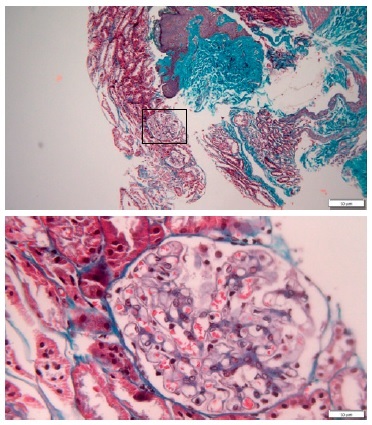
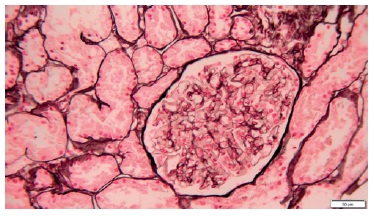
A percutaneous ultrasound-guided kidney biopsy was performed. On light microscopy, only a slightly diffused thickened glomerular basement membrane throughout all glomeruli was apparent (Figs. 2 and 3). The routine immunofluorescence of a frozen specimen containing nine glomeruli showed fine granular capillary wall deposits of polyclonal immunoglobulin G (3+), C3c (1+), kappa (1+), lambda (1+), and IgM (+/-). Renal tissue staining for PLA2R was negative. Unfortunately, images of the immunofluorescence microscopy are not available and electron microscopy was not performed. These changes in renal morphology and immunohistochemistry were consistent with stage 1 membranous nephropathy.
Initially, antinuclear antibodies (ANA) were positive (titer, 1:160) and the extractable nuclear antigen (ENAs) panel showed positivity for Scl-70 antibody (3+). A decrease in titers was seen in monthly follow-up measurements (ANA titers stabilized in 1:80 and Scl-70 decreased progressively to become negative after six months).
Anti-double stranded DNA (dsDNA) and anti-nucleosome antibodies were always negative. From the remaining study it is worth mentioning that Epstein-Barr virus (EBV) serology showed positive viral capsid antigen IgM (VCA IgM, titer 58 U/mL [positive >40]); positive VCA IgG (VCA IgG, titer 617 U/mL [positive >25]); negative anti-early antigen IgG (EA IgG, titer <5 U/mL [positive >40]); and positive nuclear antigen IgG (EBNA, titer 189 U/mL [positive >20]). After four months, VCA IgM decreased to 30 U/mL; VCA IgG titers increased to 718 U/mL; EA IgG remained negative <5 U/mL; and EBNA IgG increased to 277 U/mL. The EBV viral load was negative in both timepoints.
Further laboratory workup was unremarkable as follows: sérum anti- PLA2R antibodies were negative; complement components were normal (CH50 70 U/mL [reference: >42]; AH 50 90% [reference: >70%]; C3 1.76 g/L [reference: 0.90-1.80]; and C4 0.36 g/L [0.10-0.40]); there was no evidence of haemolysis; antistreptolysin O (72 UI/mL [reference: 0-200]) and anti-deoxyribonuclease B titres (<76 U/L [reference: 0-200]) were normal; serum amyloid protein A (4.89 mg/L [reference: <6.4] and serum electrophoresis were normal; serologies for hepatitis B virus, hepatitis C virus, human immunodeficiency virus, cytomegalovirus, parvovirus, and toxoplasmosis were negative; venereal disease research laboratory (VDRL) was negative; and interferon-gamma release assay (IGRA) was negative. Blood and urine cultures were also negative. Chest radiograph showed no pleural or parenchymal alterations.
Abdominal ultrasound showed unmodified changes related to Wilson’s disease (mild increased liver echogenicity) and the kidneys were normal. In the following two weeks the patient’s condition worsened.
Despite serum albumin remained relatively unchanged in the range of 26.5-27.5 g/L, proteinuria continued to increase reaching the nephrotic range (maximum proteinuria of 1656 mg in 12 hours or 75mg/m2/h) with associated increasing oedema and additional weight gain to a maximum weight of 74 kg. Therefore, the patient was started on oral prednisolone at 60 mg daily for four weeks (approximately 30 mg/m2/day), followed by 60 mg on alternate days for another four weeks, and slow tapering until discontinuation after a total of six months of steroids. Gradual improvement was noted, with resolution of the oedema in one month, normalization of serum albumin in two months, and complete resolution of proteinuria in five months.
Presently, at one-year of follow-up after the nephrotic syndrome onset, medications other than zinc acetate were stopped and the patient remains asymptomatic, with normal blood pressure, liver function tests, cooper biochemistry, renal function, serum albumin, lipid profile, urinalysis and urinary sediment.
DISCUSSION
Wilson’s disease is a rare autosomal recessive disorder of cooper metabolism, with an estimated prevalence of 1:30 000.9 It is caused by pathogenic mutations in the ATP7B gene, which encodes a copper transporting P-type ATPase. This protein is required for copper incorporation into ceruloplasmin and for biliary copper excretion.
Therefore, Wilson’s disease is characterized by tissular cooper overload despite low circulating levels of ceruloplasmin (the major form of circulating copper). Progressive toxic copper accumulation occurs primarily in the hepatocytes.10 Over time, cooper deposition also affects the central nervous system, cornea, myocardium, and renal proximal tubular epithelial cells. If left untreated, the disease may progress to liver failure and irreversible neurologic impairment. In contrast, early and adequate life-long treatment allows for excelente prognosis. Treatment consists of an initial decoppering phase aimed at removing cooper excess, followed by a maintenance phase aimed at preventing copper reaccumulation. Although high-quality evidence is lacking to support the first treatment choice, penicillamine remains the standard initial treatment for symptomatic patients with Wilson’s disease.7,11
The free sulfhydryl group in penicillamine acts as a cooper chelating moiety, promoting its urinary excretion. Despite being highly effective, penicillamine is associated with significant adverse effects resulting in drug withdrawal in 5% to 30% of patients. In the first three weeks after therapy initiation early sensitivity reactions may present with fever, cutaneous eruptions, neutropenia, thrombocytopenia, lymphadenopathy, and proteinuria. Other potential adverse effects may occur from months to years, which include: nausea, vomiting, and anorexia (usually resolve with dose reduction); thrombocytopenia, neutropenia, anaemia, or bone marrow toxicity with severe aplasia (which is rare but may not resolve after therapy discontinuation); anticollagen effects, such as elastosis perforans serpinginosa, cutis laxa, pemphigus, lichen planus, and aphthous stomatitis; proteinuria; isolated elevations in serum antinuclear antibodies and drug-induced lupus-like syndrome.12,13Therefore, proteinuria may appear at any time during treatment and patients treated with penicillamine should be regularly monitored for proteinuria. As part of the screening for penicillamine related adverse effects, a complete blood count, serum creatinine, and urinalysis should be obtained in the first week and monthly for the first three months of therapy, followed by three-monthly evaluations until stabilization, and semesterly thereafter.
Proteinuria occurs in 4%-33% of patients treated with penicillamine. Although it may be transient, in 60%-70% of patients who develop proteinuria it may progress to overt nephrotic syndrome.14
In a review of 63 cases of nephrotic syndrome associated with penicillamine, the mean duration of treatment was 7.6 (±3.9) months at the onset of proteinuria and 11.9 (±18.8) months at the onset of nephrotic syndrome. Accordingly, the nephrotic syndrome was diagnosed on average five months after proteinuria was first noticed.15
In our patient, the first evidence of proteinuria appeared at five months after penicillamine initiation. In the literature, the attitude towards incident proteinuria is not consensual. While some authors would keep penicillamine if quantitative proteinuria is stable and does not exceed 1 g per day, others would discontinue it if proteinuria exceeds 2+ on a dipstick or in the presence of haematuria. A consensos exists for drug withdrawal when patients present with decreasing glomerular filtration rate or when proteinuria exceeds 2g per day.16,17 In the case we reported, as the urinalysis showed isolated and mild proteinuria (50 mg/dL), a decision to keep treatment under close observation was initially made, considering that the patient was otherwise well and showed a very effective clinical and laboratorial response to Wilson’s disease treatment. We acknowledge that having used a quantitative method to measure proteinuria when it first appeared may have allowed for a timely identification of rising proteinuria before the onset of overt nephrotic syndrome.
It should be noted, however, that this occurred during the second peak of the COVID-19 pandemics in Portugal, which contributed to increase time between visits and laboratorial assessments in the outpatient clinics.
We believe this case illustrates that alternative therapies to penicillamine with a better safety profile are needed. Trientine is also an effective cooper chelating agent, with a similar although less frequent profile of adverse effects. Being more expensive, trientine has been used as a second-line option reserved for symptomatic patients who are intolerant to penicillamine. Although there is a lack of robust studies comparing trientine to penicillamine, trientine was associated with a higher risk of worsening neurologic symptoms after therapy initiation. As such, it was not considered a good option in our patient who presented initially with liver and neuropsychiatric disease. Our decision was to substitute penicillamine for zinc acetate, taking into consideration that Wilson’s disease was adequately controlled. Zinc salts activate metallothionein in enterocytes reducing dietary copper absorption. Given its safety profile, zinc salts are used as first-line therapy for presymptomatic patients with Wilson’s disease identified through family screening and as life-long maintenance therapy for stabilized patients after the initial decoopering phase of treatment. In general, the recommended initial treatment for symptomatic patients with Wilson’s disease is a copper chelating agent, such as penicillamine or trientine, followed by a maintenance phase with a lower dose of a chelating agent, zinc salts, or a combination of both. As limited data are available to guide the decision on when to switch therapy, the strategy should be individualized according to the type and severity of organ involvement. The time required to achieve clinical and biochemical control under higher-dose chelating therapy to allow for transition to the maintenance phase is highly variable, from six months to five years. Therefore, patients treated initially with penicillamine should be followed-up closely for compliance, effectiveness and adverse effects. The occurrence of penicillamine related adverse effects should prompt drug discontinuation and switching to trientine or zinc salts according to the severity of the disease.7,11
Our patient renal biopsy was consistent with membranous nephropathy. It is the most frequent morphologic pattern of injury associated with penicillamine exposure, occurring in 55% of cases, followed by minimal change disease in 27%. Other renal histologic findings have also been described in patients treated with penicillamine, although less frequently, such as IgM nephropathy (4%), mesangioproliferative glomerulonephritis (2%), membranoproliferative glomerulonephritis (2%), and focal interstitial nephritis (2%).14,15,18,19A few cases of ANCA-associated glomerulonephritis have also been reported in patients treated with penicillamine for rheumatoid arthritis or systemic sclerosis, but it has also been described in patients presenting with these disorders in the absence of treatment with penicillamine. There is, however, a case report of a female adolescent treated with penicillamine for Wilson’s disease who presented with rapidly progressive ANCA-associated crescentic glomerulonephritis.20 Although Wilson’s disease itself may be associated with tubular dysfunction due to cooper deposition, glomerulopathy is rare. In contrast, penicillamine has long been associated with glomerular disease. The mechanisms of penicillamine-induced renal damage are essentially unknown. Nevertheless, the development of proteinuria after penicillamine initiation, its improvement after discontinuation, and its recurrence after the drug reintroduction support a causal role as a nephrotoxic drug.
Moreover, negativity for anti-PLA2R in serum and for PLA2R staining in renal tissue supports that membranous nephropathy in our case was secondary. Likewise, indirect immunofluorescence positivity for reactants beyond C3 and IgG also supports this diagnosis. Low ANA titers are frequent in the general population of female adolescentes and titers may increase transiently during unrelated acute illnesses, which is likely to have been the case in our patient. In the absence of other clinical and laboratorial markers for either diffuse type of systemic sclerosis and systemic lupus erythematosus, transient Scl-70 positivity was also interpreted as either an epiphenomenon or as a result of drug exposure.21 Its disappearance over the course of the disease also supports this hypothesis, although we will keep monitoring for further evidence of pathological autoimmunity. The EBV serology suggests that a recent reactivation of infection may have played a role as a trigger for the nephrotic syndrome and other imune mediated events. An EBV reactivation as the direct cause of the clinical presentation is less plausible considering all the topics already discussed and the negative viral load.
Proteinuria is known to resolve or at least show a significant reduction to less than 1 g in 24 hours in about 7.1 (±5.9) months after discontinuation of penicillamine. It is also known that it may further increase over one to four months after discontinuation of penicillamine in nearly half the patients before it starts to decline consistently.
Despite knowing that, we treated our patient with steroids in light of the rapidly increasing oedema and weight gain due to increasing nephrotic range proteinuria. Observational studies have suggested that recovery is faster when steroids are used. The overall prognosis is good, with associated decreased glomerular filtration rate being na exception rather than a rule.14,15
Finally, we aim to emphasize that the onset of proteinuria in patients treated with penicillamine may herald the progression to nephrotic syndrome. Whenever proteinuria is suspected, it should be measured using a quantitative method. The presence of significant proteinuria in these patient population should prompt the multidisciplinar discussion about penicillamine discontinuation or substitution for an alternative drug.

