











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
X-linked hypophosphatemic rickets (XLHR) is a rare, multisystemic disorder, which can cause lifelong disability.1 It has been linked to loss-of-function PHEX (phosphate-regulating endopeptidase homolog, X-linked) mutations, which increase fibroblast growth factor 23 (FGF23) levels. FGF23 inhibits phosphate reabsorption in the renal tubules, causing renal phosphate wasting. In parallel, FGF23 blocks the synthesis of the active form of vitamin D, reducing phosphate gut absorption and contributing to the hypophosphatemia characteristic of XLHR. A decrease in phosphate levels compromises bone mineralization, causing rickets and osteomalacia.2
Other clinical manifestations of XLHR include short stature, boné deformities, gait abnormalities, dental anomalies, hearing loss, craniosynostosis and Arnold-Chiari malformations. Patients may also presente osteoarticular pain, enthesopathy, fractures, pseudofractures and spinal stenosis.2,3
Until recently, XLHR treatment consisted of supplements of phosphate and active vitamin D analogues, and orthopedic surgery, if needed to correct limb deformities. However, conventional therapy is limited by difficult compliance, as well as renal (hypercalciuria, nephrocalcinosis, nephrolithiasis), gastrointestinal (abdominal pain and diarrhea) and endocrinological (hyperparathyroidism) side effects. In addition, some patients may have an insufficient response to treatment.4 In 2018, a human recombinant IgG1 monoclonal antibody anti-FGF23, burosumab, was approved by the European Medicines Agency and the Food and Drug Administration for the treatment of XLHR.5,6 Burosumab has shown promising results, as it has been associated with improvement of rickets to a greater extent than conventional therapy.7-9
We here report a XLHR patient, who is illustrative of the disease as the patient presented with bone deformities and complications such as an Arnold-Chiari malformation, as well as side effects from conventional therapy, namely nephrocalcinosis, hyperparathyroidism and diarrhea. With an early start of burosumab in a young patient, our hope is to change the patient’s prognosis and minimize complications.
CASE REPORT
We present a case of a 4-year-old female, without family history of XLHR. Pregnancy, delivery and neonatal period were unremarkable.
The birth length was 50 cm (50th percentile). At 12 months the patient’s mother detected deformities in the lower limbs and an abnormal gait. These changes were initially interpreted by the pediatrician and na orthopedist as a normal variant. However, due to worsening of the deformities, at 23 months, laboratory tests were performed. It was detected hypophosphatemia (2.5 mg/dL, reference range for age: 3.8-6.5 mg/dL), normal vitamin D (23.6 ng/mL), calcium (9.7 mg/dL) and parathyroid hormone (PTH) (42.4 mg/dL) levels (Table 1). A diagnosis of XLHR was suspected and the patient initiated conventional therapy with 250 mg oral phosphorus three times a day (750 mg/day; 73 mg/kg/day) and 1334 IU of cholecalciferol a day.
Table 1 Biochemical analysis of the patient throughout the history of the illness

Y: years; m: months; PTH: parathyroid hormone; ALP: alkaline phosphatase; TRP: tubular reabsorption of phosphate; TmP/GFR: tubular maximum reabsorption of phosphate corrected for glomerular filtration rate.
At the age of 3, genetic testing identified an heterozygotic deletion on exon 10 of PHEX, confirming XLHR diagnosis. The parents and younger brother did not have any changes on PHEX, suggesting a de novo mutation in the patient.
Oral phosphate supplementation was increased until 1500 mg/day (132 mg/kg/day) and the patient had frequent diarrhea. While the patient was under conventional therapy, her phosphate levels were below reference range (1.7 mg/dL, reference range for age: 3.7-5.6 mg/dL), while PTH and alkaline phosphatase (ALP) levels were above reference range (81.6 pg/mL and 817 U/L, respectively). Vitamin D and calcium levels were normal (31.3 ng/mL and 9.8 mg/dL) (Table 1). A renal ultrasound performed at 3 years old identified medullary nephrocalcinosis stage II/III (Fig. 1).
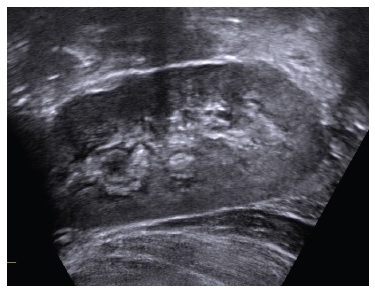
Figure 1 Renal ultrasound - hyperechogenicity on renal pyramids, secondary to medullary nephrocalcinosis.
The patient began being followed by our team when she turned 4 years old. At this time, the patient presented bone deformities, frequent nonspecific musculoskeletal pain and fatigue, as well as side effects from conventional therapy, namely nephrocalcinosis, hyperparathyroidism and frequent diarrhea. The renal ultrasound was repeated and showed worsening of the nephrocalcinosis. An Arnold-Chiari type I malformation was identified by brain magnetic resonance (Fig. 2). The patient denied headaches and vomiting. Focal signs, impaired cranial nerves, breathing irregularities and cranial shape or size alteration were absent.

Figure 2 Brain magnetic resonance - low position of the cerebellar tonsils, congruent with an Arnold-Chiari type I malformation.
At observation, the patient’s weight was 18.5 kg (75th percentile) and height was 101.2 cm (25th percentile). The patient’s target height, as predicted by parental heights (mother’s height: 156 cm, father’s height: 178 cm), is between percentiles 25th and 50th. The patient’s weight and height have been stable throughout the patient’s life. The head circumference was 52.2 cm (95th percentile). BMI was 17.6 kg/m2 (between the 90th and 95th percentiles) (Fig. 3). A bilateral varus deformity of lower limbs was present (Fig. 4); as well as metaphyseal widening at the wrists and ankles (Fig. 4). Rachitic rosary and Harrison’s groove were absent. The patient never had dental abscesses or other dental abnormalities.
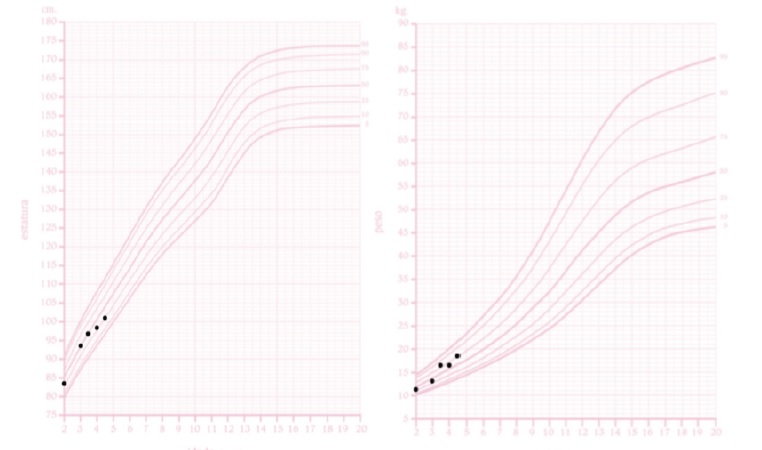
Figure 3 The patient’s height for age curve (left) and weight for age curve (right), for girls, according to the Portuguese General Health Department. The x-axis represents the age in years while the y-axis corresponds to the length in centimeters (left) and weight in kilograms (right).

Figure 4 A: Bilateral varus deformity of lower limbs with metaphyseal widening at the wrist and ankle level (left and right). B: Bilateral varus deformity of lower limbs in radiographs (left and right).
Conventional therapy was stopped and burosumab was initiated, with a dose of 20 mg every 2 weeks (1.1 mg/kg). After only one dose of burosumab, we observed an increase in serum fasting phosphate levels (3.6 mg/dL), normalization of PTH levels (61.5 pg/mL), and a decrease in ALP levels (670 U/L). After 2 months of burosumab, phosphate levels were 3.1 mg/dL, total vitamin D, calcium and PTH levels were normal and ALP levels decreased to 372 U/L. After 5 months of burosumab, phosphate levels were normal (3.8 mg/dL), total vitamin D, calcium and PTH levels remained normal, and ALP levels were 493 U/L. No side effects were identified. The evolution of the bone deformities and of the nephrocalcinosis will be evaluated by imaging one year after the start of burosumab.
DISCUSSION
XLHR is an heterogeneous disease, characterized by a wide variety of manifestations of different severities.10,11 In this child, the diagnosis was initially established based on clinical, biochemical and radiological findings, and was later confirmed by genetic testing.12
The clinical presentation is similar to what was previously described,13,14 with a varus deformity of the lower extremities observed after the onset of walking, as well as an unusual gait. The first doctors who evaluated the patient did not investigate an underlying cause and considered it to be a normal variant. Diagnostic delays are frequent in this disease.13,15
XLHR patients tendentially have normal stature at birth, but, progressively, develop short stature with relatively preserved trunk growth.1 This patient maintained a stable height on the expected percentile according to her parents’ heights.
XLHR has been linked to musculoskeletal symptoms that lead to mobility impairment and reduced physical activity, resulting in a higher risk of obesity.16 As per her BMI, the patient is considered to be overweight.
She suffered from frequent nonspecific musculoskeletal pain, but she still remained physically active. The patient has not taken a 6 minute walk test that would help assess musculoskeletal involvement in XLHR, as she is not yet 5 years old, the age recommended to begin screening.4
Hypophosphatemia is the main biochemical feature of XLHR. Normal plasma phosphate levels vary with age, with higher values observed in infants.1 Furthermore, phosphate levels follow a circadian variation and are also influenced by diet.18 The patient presented hypophosphatemia at the time of diagnosis and during conventional therapy. In XLHR, hypophosphatemia is secondary to renal phosphate wasting, so the tubular maximum reabsorption of phosphate corrected for glomerular filtration rate (TmP/GFR) is usually decreased. TmP/GFR values vary with age.19 In this patient, TmP/GFR was below the reference range for age before the start of burosumab, showing renal phosphate wasting.
ALP is a biomarker of rickets activity and thus of treatment efficacy.15 Total ALP can be used to monitor children with XLHR, as bonespecific ALP represents 80% to 90% of total ALP in this age group. In adults, 50% of total ALP levels originate from the liver and, thus, bone-specific ALP is preferred to monitor XLHR patients.4 In this patient, ALP levels were increased, even with conventional treatment.
With the start of burosumab, ALP levels progressively reduced. As ALP is the parameter that determines rickets activity, a reduction in its levels is one of the goals of XLHR treatment.
Vitamin D and calcium plasma levels were always within normal range. This helps differentiate XLHR from rickets secondary to a disorder of calcium metabolism.12 PTH levels were normal at presentation, increased to above normal range with conventional therapy, and were again normal after the start of burosumab. This is expected, as conventional therapy has been linked to hyperparathyroidism and this does not occur with burosumab.7-9 Moreover, the patient developed other side effects of conventional therapy, such as diarrhea and nephrocalcinosis.
FGF23 levels tend to be elevated, but normal levels do not exclude XLHR.4 FGF23 measurement was not available at our institution. In addition to clinical and biochemical features, radiographic findings are crucial to assess the severity of rickets. The radiographic findings are best visualized at growth plates in sites of rapid growth, namely, the radio and ulna, tibia, distal femur, and costochondral junctions.20 Rachitic lesions are characterized by loss of definition, widening and irregular growth plates, along with cupped, widened or frayed metaphyses,4 as we could see in our patient.
Burosumab is a monoclonal antibody IgG1 against FGF23 that directly targets the underlying disease mechanism. In a phase 2 clinical trial of children with XLHR aged 1 to 4 years, burosumab had a favorable safety profile and significantly increased serum phosphate, improved rickets and prevented early declines in growth.7 In another, larger, phase 2 clinical trial of children with XLHR, aged 5 to 12 years, treatment with burosumab improved renal tubular phosphate reabsorption, serum phosphate levels, linear growth and physical function, and reduced pain and rickets severity.8 Finally, a phase 3 efficacy and safety trial among children with XLHR aged 1 to 12 years showed that patients who switched from conventional therapy to burosumab had a greater improvement of rickets severity and growth.9 Furthermore, burosumab has not been linked to the complications of conventional therapy, such as nephrocalcinosis and hyperparathyroidism,7-9 a positive factor for XLHR patients who frequently develop these complications, as was seen in this patient.
As XLHR is heterogeneous in clinical presentation and severity, it is also variable in its response to treatment. So far, it has not been possible to predict how the disease will respond, and who would benefit most from treatment.
This is a didactic case of XLHR, as the patient presented with lower limb deformities that were initially considered to be normal, and developed serious side effects secondary to conventional therapy.
Moreover, the patient had an asymptomatic Arnold Chiari malformation, which had not previously been detected. Taking into account our patient’s age and growth potential, she is a preferential target to try to prevent short stature and bone deformities caused by this disease.
It is fundamental to recognize the disease in its early stages and to learn about its possible new form of treatment.
XLHR diagnosis tends to be hampered by its rarity and wide intra- and interfamilial phenotypic diversity. Being a rare condition where early treatment allows for a better control of the disease, physician’s awareness must be increased to enable rapid disease recognition and diagnosis. Furthermore, XLHR is a multisystemic disease that requires a multidisciplinar approach to correctly manage and control its manifestations.
Although the biochemical evolution of our patient has been positive since the start of burosumab, it is still too early to determine its full impact and potential benefits; therefore, longer time under burosumab therapy is needed.
Further studies should focus on predictors of treatment response and evaluating the impact of burosumab in the quality of life of patients with XLHR. It is also important to evaluate the cost-benefit of this treatment, when to stop the drug administration and the benefits and disadvantages of its use on adult patients with XLHR. By bringing this clinical case to light, our aim is to discuss the disease manifestations and complications to allow for a rapid diagnosis, as well as to highlight the possibility of a new treatment strategy.
More and longer clinical trials are needed to guarantee efficacy, safety and cost-effectiveness. We hope the current positive biochemical evolution of our patient, who began this treatment at a young age, will possibly minimize the major complications of XLHR.17

