











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Focal segmental glomerulosclerosis (FSGS) is a kidney histological lesion, frequently identified in children and adults presenting with nephrotic syndrome. It can be subdivided in primary, secondary, genetic and unknown forms, according to the underlying pathological mechanism. The histological identification of a FSGS lesion should not be the end of the diagnostic work-up, but rather the trigger to identify the underlying causative mechanism, since this has major therapeutic and prognostic implications. We present the case of a patient with genetic FSGS caused by a rare combination of two variants in the NPHS2 gene.
CASE REPORT
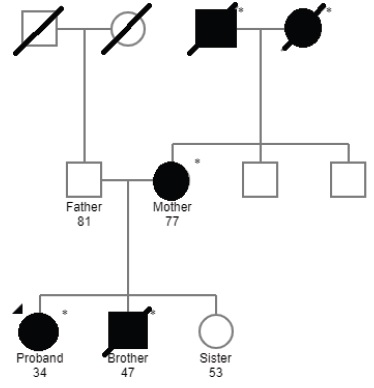
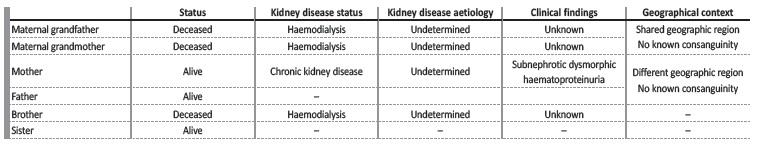
We describe the case of a 34-year-old white female, with previous medical history of pre-eclampsia during the first pregnancy (at the age of 20-years old), psoriasis and family history of kidney disease of undetermined aetiology (both maternal grandparents and brother on haemodialysis, mother with chronic kidney disease, no consanguinity.
See Fig. 1 and Table 1 for more information). She was evaluated by a general practitioner in October 2019, where a diagnosis of arterial hypertension was established and she was started on losartan 100 mg per day. One month later, she reported foamy urine and a dipstick test was performed, revealing the presence of proteins and blood in the urine. Sediment analysis revealed dysmorphic red blood cells in the urine. For this reason, she was referred to our Nephrology unit. The patient denied other complaints such as oedema, macroscopic haematuria, polyuria, lumbar pain, anorexia, changes in body weight, consumption of non-steroidal anti-inflammatory drugs, antibiotics or other drugs other than losartan. No extra-renal disease manifestations were identifiable.
INVESTIGATIONS
The relevant bloodwork results are shown in Table 2. We highlight the analytical findings of microscopic haematuria, nephrotic-range proteinuria and concomitant hypoalbuminaemia. Chest X-ray was unremarkable. Kidney ultrasound showed normalsized kidneys with maintained parenchymal thickness and corticomedullary differentiation. Further studies were requested in order to investigate the aetiology of the nephrotic syndrome. ANA, ANCA, anti-soluble nuclear antigen antibodies, anti-dsDNA antibodies and anti-phospholipase A2 receptor (anti-PLA2r) antibodies were negative. Immunoglobulins, serum free light chains and respective ratio, serum protein electrophoresis and complement levels were normal. Infection-related serologies were also performed, with a negative result for hepatitis B and C and human immunodeficiency viruses 1 and 2. Interferon gamma release assay (IGRA) was positive, but the patient did not present any symptoms related to mycobacterial infection and, thus, it was assumed as latent tuberculosis.
Table 2: Relevant bloodwork results in the initial Nephrology evaluation.
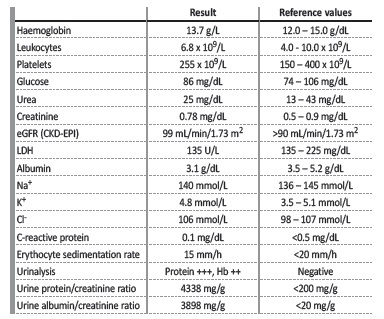
Hb - haemoglobin; LDH - lactate dehydrogenase.
The patient was submitted to a kidney biopsy. In light microscopy (Figs. 2 and 3), 20 glomeruli were observed, two of which presented lesions of focal segmental glomerulosclerosis (FSGS) with not otherwise specified pattern. The remaining glomeruli did not present any lesions. Proximal and distal tubule cells had protein inclusions in the cytoplasm. Interstitial fibrosis and tubular atrophy were observed in under 10% of the sample. Immunofluorescence study was negative for Immunoglobulins A, G and M, C1, C3, albumin and kappa/lambda light chains. Electron microscopy highlighted podocyte hyperplasia and foot process effacement affecting approximately 50% of the glomerular tuft (Fig. 4).
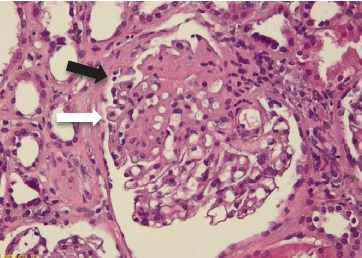
Figure 2: Light microscopy, 10x, haematoxylin and eosin: segmental thickening of the tuft, highlighting rare xanthomatous endocapillary cells (white arrow) and hint of segmental lesion adhering to Bowman’s capsule (synechia) (black arrow)
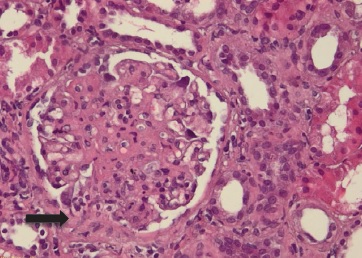
Figure 3: Light microscopy, 10x, haematoxylin and eosin: segmental tuft thickening and synechia (black arrow).
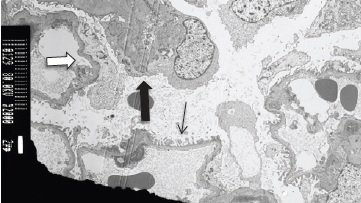
Figure 4: Electron microscopy, 2000x: portion of the glomerular tuft highlighting segmental foot process effacement in an area of preserved basal membrane (white arrow) and in an area of a collapsed capillary loop (black thick arrow). Foot process effacement was evident in 50% of the glomerular tuft. Areas of foot process preservation were also observed (black thin arrow).
The findings in our patient, namely the age of onset, absence of known risk factors for secondary FSGS and histological examination, FSGS. Additionally, the patient had a family history of kidney disease of unknown cause. Thus, genetic testing was requested. Genetic study was performed at Genomed laboratory after extraction of DNA from peripheral blood and analysis was conducted using “Twist human core exome plus RefSeq extension” kit (Twist Bioscience) followed by parallel massive sequencing [Next generation sequencing (NGS)] and confirmation by Sanger sequencing for regions not accurately analyzed by NGS. The study showed two variants in the NPHS2 gene: c.686G>A, p.(Arg229Gln), which has been reported as a risk factor for kidney disease when combined with another pathogenic variant in the same gene1; c.855_856del, p(Arg286Thrfs*17), a rare variant described as pathogenic when present in compound heterozygosity with the former c.686G>A, p.(Arg229Gln) mutation.2 Mutations occurring in the NPHS2 gene are associated with steroid-resistant nephrotic syndrome (SRNS) (OMIM 600995) with an autosomal recessive inheritance, compatible with the diagnosis of genetic FSGS.
TREATMENT, OUTCOME AND FOLLOW-UP
While waiting on the results of electron microscopy and genetic testing, given the clinical picture and the light microscopy findings, the patient was started on corticosteroids (prednisolone 1 mg/kg/day), after adequate immunizations, and also tuberculosis prophylaxis with isoniazid and pyridoxine, due to positive IGRA. No improvement was recorded after initiation of therapy, and, additionally, during this period, the result of the genetic test and electron microscopy was made available. Thus, after 9 weeks of treatment she started tapering and later suspended corticosteroids. After withdrawal of prednisolone, proteinuria decreased to 3739 mg/day and serum albumin rose to 3.7 g/dL. Taking into account the diagnosis, additional courses of other immunosuppressive agents were not initiated. The patient is currently 37-years old, still under regular Nephrology follow-up, three years after the referral, maintaining a serum creatinine of 0.8 mg/dL, proteinuria of 3500-4000 mg/day, serum albumin >3.5 g/dL and controlled arterial hypertension, without any other abnormalities. Additionally, she is on supportive treatment with losartan and dietary recommendations since the beginning of the follow-up.
DISCUSSION
The differential diagnosis of nephrotic-range proteinuria, with or without associated nephrotic syndrome, is broad, and, therefore, a systematic approach is recommended. In our patient, the absence of symptoms suggestive of systemic disease, such as systemic lupus erythematosus, diabetes mellitus or amyloidosis, which are responsible for approximately 30% of nephrotic syndromes observed in adults,3 pointed towards a primary kidney disorder. The most frequent primary kidney diseases responsible for nephrotic syndrome in adults are minimal change disease, FSGS and membranous nephropathy, although other entities (IgA nephropathy or infection-associated glomerulonephritis) can seldom present with a nephrotic phenotype.4 The absence of serum anti-PLA2r antibodies reduced the degree of suspicion for membranous nephropathy, although it could not be excluded. In cases of suspected podocytopathy, histological examination is necessary to further elucidate about the underlying pathological mechanism. The FSGS lesions documented in light microscopy, in the context of the presented clinical picture, provided enough evidence to establish the diagnosis of FSGS, but that is not the final step in the diagnostic evaluation of such patients.
Differential diagnosis between different forms of FSGS is importante for therapeutic and prognostic reasons, and may be aided by electron microscopy and clinical/analytical abnormalities. The findings on light microscopy do not allow the diagnosis of a particular form of FSGS, but the evaluation of the extent of podocyte foot process effacement is possible with electron microscopy.5 While this lesion is diffuse in primary FSGS, it tends to be more limited in secondary forms of FSGS.6
Also, the clinical presentation of patients tends to differ between primary and non-primary forms of FSGS, with the former presenting more frequently with full-blown nephrotic syndrome and marked proteinuria, while secondary FSGS is more often suggested by progressively increasing proteinuria and declining kidney function.7,8 The clinical presentation in patients with genetic FSGS is widely variable, as it may manifest in early infancy with nephrotic syndrome or later in adulthood with progressive chronic kidney disease and less severe proteinuria.5
Most patients with childhood-onset genetic FSGS have autosomal recessive mutations with full penetrance, commonly presenting with nephrotic syndrome.9 Adult-onset genetic FSGS is more frequently associated with autosomal dominant inheritance and variable penetrance,10 as suggested by the family pedigree (see Fig. 1). In our patient, two variants were identified in the NPHS2 gene:
• c.686G>A, p.(Arg229Gln), identified in exon 5, which has been reported as a risk factor for kidney disease when combined with a pathogenic variant in exons 7 and 8 of the NPHS2.1,11 This single-nucleotide polymorphism causes a missense protein and is frequent in population databases [ClinVar - ID: 5370, Human Gene Mutation Database (HGMD); Minor allele frequency (MAF) in Genome Aggregation Database (GnomAD) of3%];
• c.855_856del, p(Arg286Thrfs*17), identified in exon 7, is a rare variant reported previously in patients with FSGS presenting with nephrotic syndrome.12-14 It is infrequent in population databases [ClinVar - ID: 188823, HGMD; MAF GnomAD of 0.0071%], only described once in compound heterozygosity with c.686G>A, p.(Arg229Gln) mutation.2 This deletion generates a premature stop codon and is classified as probably pathogenic [American College of Medical Genetics and Genomics (ACMG) 2015/Association for Clinical Genomic Science (ACGS) 2019: PVS1_str, PM1, PM2, PM3_sup].
The NPHS2 gene (OMIM 604766) codifies for podocin, a transmembrane protein found exclusively in glomerular podocytes, important in the recruitment of nephrin to the slit diaphragm, firstly identified in families with autosomal recessive steroid-resistant nephrotic syndrome (SRNS).15 NPHS2 mutations are usually associated with early onset of disease and progression to end-stage kidney disease (ESKD) within the first decade,14 but our patient did not present any symptoms or abnormalities during infancy or adolescence. A paper from the Mayo Clinic reported two siblings with the same compound heterozygote mutation as our patient. Despite sharing the same genotype, one presented with full nephrotic syndrome and diffuse foot process effacement, while the other presented with subnephrotic proteinuria with only partial foot process effacement. This may be due to the intricate interaction between genotype, environment and certain epigenetic phenomena, leading to drastically different phenotypes.16
Resistance to glucocorticoid therapy, while also possible in primary FSGS, is frequent in patients with monogenic forms of FSGS, particularly children.5 Glucocorticoid resistance in adults with genetic FSGS is less certain, and a small subgroup may respond to therapy, even if lasting complete remissions are scarce.17 Mutations in the NPHS2 gene are usually resistant to immunosuppression, but there are reports of response in heterozygous mutations.5 Our patient was initiated on corticosteroid treatment due to the findings of FSGS lesions in light microscopy while waiting on the result of electron microscopy and genetic testing. Taking into account the results of both studies and the lack of response to therapy, we suspended the corticosteroids to avoid iatrogenic complications. There are other treatment options used for primary FSGS, such as calcineurin inhibitors, mycophenolate mofetil or rituximab, but response to immunosuppression in patients with genetic FSGS is rare.5 These were discussed with the patient, but taking into account the potential adverse effects of these agents, the absence of nephrotic syndrome and the profound immunosuppression during the coronavirus pandemic, no additional therapeutic agentes were initiated.
Another factor to consider is the prognostic importance of genetic testing and counseling in genetic FSGS identifying a genetic cause for FSGS has a tremendous impact in the patient’s management, as it not only allows the avoidance of the toxicity of immunosuppression, but also affects the evaluation for kidney transplantation. In this matter, genetic FSGS compares favourably to primary FSGS, since the former recurs in 0%-8% and the latter in 30%-70% of patients.18-21 Additionally, there are reports suggesting a good prognosis in patients with genetic FSGS caused by NPHS2 mutations.21 Identifying a genetic form of FSGS is also important for living donation, as it should prompt a screening of family members who are potential donors, since there are reported cases of FSGS development in the donors’ remaining kidney.22
CONCLUSION
We describe a case of genetic FSGS caused by a rare combination of NPHS2 variants in compound heterozygosity, providing further insight about the diagnostic evaluation of patients with FSGS lesions on kidney biopsy. Although genetic FSGS is not a frequent diagnosis, it should be suspected in individuals with nephrotic-range proteinuria/nephrotic syndrome, family history of kidney disease and FSGS lesions in kidney biopsy, especially if resistant to immunosuppression.
Although patients with NPHS2 gene variants are usually resistant to immunosuppression, kidney transplant appears to be a good option with a low rate of recurrence.

