











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Primary hyperoxaluria type 1 (PH1) is a rare autosomal recessive disease caused by mutations in the AGXT gene, leading to a deficiency of the liver peroxisomal enzyme alanine:glyoxylate-aminotransferase (AGT), which catalyzes the conversion of glyoxalate to glycine.1 This deficiency results in accumulation of glyoxalate and overproduction of oxalate and glycolate. Oxalate and calcium, combined, form highly insoluble calcium oxalate salts, leading to urolithiasis, nephrocalcinosis and progressive chronic kidney disease. Clinical spectrum varies widely: recurrent nephrolithiasis and progressive chronic kidney disease (CKD) in childhood, adolescence or early adulthood (the majority of patients); infantile oxalosis characterized by nephrocalcinosis and renal failure in infancy or early childhood (~ 10%); recurrent oxalate nephropathy in a kidney allograft after transplantation (10%); occasional kidney stone disease in adulthood (<10%); or screening due to family history (<10%).2-4
Systemic oxalosis, with accumulation of calcium oxalate crystals in extra-renal tissues, may occur when estimated glomerular filtration rate (eGFR) falls below 30 - 50 mL/min per 1.73 m2, and is associated with high morbidity and mortality.5
Diagnosis of PH1 is based on measurements of urinary levels of oxalate. A persistently elevated excretion (> 45 mg/1.73 m2/day or urinary oxalate:creatinine ratio greater than the reference value for age) and suggestive clinical symptoms without a secondary cause of hyperoxaluria (dietetic or hyperabsortive), are indications for further evaluation. A definitive diagnosis requires genetic testing of AGXT gene or liver biopsy to measure AGT catalytic activity.2,5
Before 2020, treatment of PH1 was primarily managed with supportive measures consisting of hyperhydration, crystallization inhibitors (such as potassium citrate) and pyridoxine. Pyridoxine is a cofactor to AGT that might reduce oxalate production by targeting AGT into the peroxisomes, increasing AGT stability and enzymatic activity or a combination of these mechanisms.6,7 This treatment will improve the prognosis only when pyridoxine-responsive mutations are presente (such as Gly170Arg and Phe152Ile).2 Patients with kidney failure often need intensive hemodialysis to reduce the risk of systemic oxalosis.2,8
Combined or sequential liver and kidney transplantation are curative options for PH1, but are associated with high morbidity and mortality.1,8,9
Recent drugs based on RNA interference (RNAi) lead to depletion of substrate for enzymes, decreasing oxalate production.8 Lumasiran is a subcutaneously administered RNAi medication which targets messenger RNA of the HAO1 gene encoding hepatic glycolate oxidase. By inhibiting glycolate oxidase production, lumasiran reduces glyoxylate substrate available for oxalate production.10 Studies have showed the efficacy and safety of lumasiran in reducing urinary oxalate excretion and plasma oxalate.10,11 We report the case of a young female with PH1 who is the first patient treated with lumasiran in Portugal.
CASE REPORT
We report the case of a 19-year-old girl with recurrent renal colic since 13 years of age and a family history of nephrolithiasis (paternal uncle, first cousin and maternal grandfather). She was admitted to our tertiary hospital at 14 years old with complaints of abdominal pain. Abdominal radiography revealed multiple bilateral kidney stones (Fig. 1A). Renal ultrasound confirmed bilateral kidney stones of 6‑12 mm of diameter and showed obstructive right hydronephrosis (35 mm of posteroanterior diameter) and ureteral dilatation upstream a 12 mm stone, associated with diffuse urothelial thickening. Laboratory workup (Table 1) revealed elevated serum creatinine (1.55 mg/dL), with eGFR (estimated by Schwartz formula) of 56 mL/min per 1.73 m2 and mild elevation of uric acid (6.0 mg/dL). A ureteral stent was placed, with improvement of hydronephrosis and eGFR. Kidney stone chemical analysis revealed the presence of calcium, oxalate and uric acid. The patient was discharged and referred to a pediatric nephrology outpatient appointment.
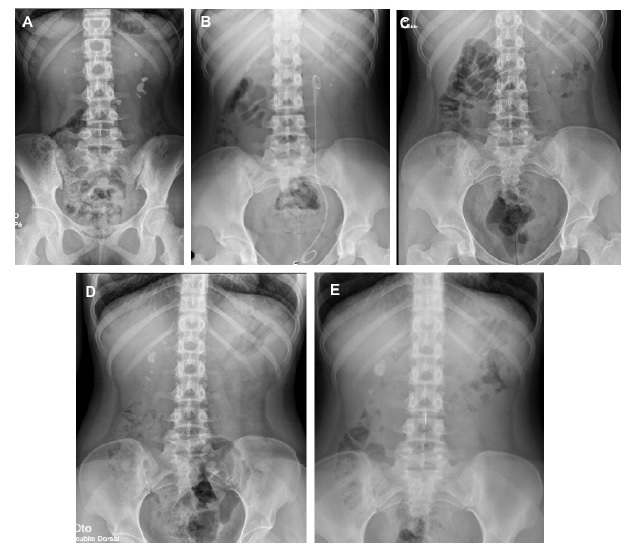
Figure 1 A. Abdominal radiography at hospital admission (14 years old); B. Radiography three months after lithotripsy (15 years old); C. Radiography 14 months after initiation of pyridoxine (16 years old); D. Radiography before initiation of lumasiran (17 years old) showing several kidney stones; E. Radiography six months after lumasiran initiation (18 years and 5 months of age) showing stability of the kidney stones.
Table 1 Laboratory work-up at the diagnosis and after initiation of conventional therapy and lumasiran
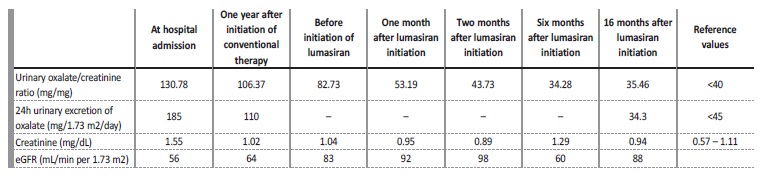
Metabolic study revealed hypocitraturia (< 0.3 mg/dL) and hyperoxaluria with high urinary oxalate/creatinine ratio of 130.78 mmol/ mol (reference value - RV: <40 mmol/mol) and 24-hour oxalate excretion of 185 mg/1.73 m2/day (RV: <45 mg/1.73 m2/day). She was started on conservative treatment with dietary oxalate restriction, hyperhydration and sodium and potassium citrate (1.7 mEq/kg/day).
During follow-up, she maintained recurrent renal colic requiring multiple surgical interventions such as extracorporeal shock wave lithotripsy and stent replacement. At the age of 14 years and 8 months, she was started on pyridoxine (initial dose of 6.3 mg/kg/day titrated up to 16.9 mg/kg/day), with irregular compliance. One year after the initiation of pyridoxine, there was a decrease of the 24-hour urinary excretion of oxalate to 110 mg/1.73 m2/day, but the patient continued to have complaints of renal colic.
At the age of 16 years, genetic testing revealed mutations in the AGXT gene, with two pathogenic variants in compound heterozygosity - c.731T>C p. (Ile244Thr) and c.1151T>C p. (Leu384Pro), confirming the diagnosis of autosomal recessive PH1. Hyperoxaluria improved (71.4 mg/1.73 m2/day) and eGFR stabilized at 83 mL/min per 1.73 m2 (stage G2 CKD), despite irregular compliance of sodium and potassium citrate and pyridoxine. However, during follow-up, she maintained renal colic and there was imagiologic worsening, with increase in the number and size of kidney stones: the larger calculi measured 20 mm and 12 mm, in the right and left kidney respectively, with no ureteral dilation (Fig. 1B-D). Cardiac evaluation was normal.
At the age of 17 years and 11 months, the patient started treatment with lumasiran (165 mg once per month for three months, followed by once every three months). She had a favorable response with rapid and sustained decrease of urinary oxalate/creatinine ratio to normal values (Table 1), no new episodes of renal colic, stability of the number and size of kidney stones and an improvement in eGFR (estimated by CKD-EPI 2009 equation) up to 92 mL/ min per 1.73 m2. There were no identifiable side effects. Currently, the patient has been under lumasiran therapy for 18 months, with no relapses.
DISCUSSION
In this case, persistent hyperoxaluria, in the absence of dietary overconsumption of oxalate, vitamin C intoxication, or intestinal malabsorption, led to the suspicion of primary hyperoxaluria, later confirmed by genetic testing. The patient had an initial irregular compliance to treatment; however, she showed partial improvement with pyridoxine. This is consistent with the genetic diagnosis, since one of the patient’s genetic variants (Ile244Thr) is known to be partially sensible to pyridoxine.6 Although the patient had a stable renal function, she maintained complaints of renal colic and there was an increase in the number and size of kidney stones during follow-up. Once lumasiran was approved in 2020, arrangements were made to initiate it.
In May 2021, at the age of 17 years and 11 months, the patient was started on this drug, which led to a rapid and consistent decrease in urinary oxalate excretion to normal levels and disappearance of monohydrate calcium oxalate crystalluria on urinary sediment microscopy.
Today, the patient has a follow-up of 18 months of treatment with lumasiran, with a stable eGFR and without new episodes of kidney stones or known side effects.
We report the case of the first patient in Portugal treated with lumasiran. Case reports of RNAi treated patients with such a prolonged follow-up are still scarce. We believe it is important to state the very favorable outcome of our patient with this therapy, and its effect on both the improvement in the quality of life and on the retardation of CKD progression. Furthermore, it is of utmost importance to understand if this drug may lead to a partial or total reduction of nephrolithiasis.
We consider that knowledge of similar cases may allow more patients to initiate lumasiran as first line therapy for PH1. We also recommend that lumasiran should be initiated at early stages of disease, to prevent additional morbidity and mortality, as well as to reduce the need for transplant in the future. Although the results are optimistic, the cost of this drug may reduce the availability to all patients.

