











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Renal hypouricemia (RHUC) is a rare hereditary disease, transmitted on an autosomal recessive pattern and is defined by the presence of low serum uric acid (UA) levels due to increased renal excretion.
This is a direct consequence of loss-of-function or inactivating mutations in one of the two main urate renal transporters located in proximal tubule cells. It is classified as type 1 or type 2 depending on the mutated transporter, urate transporter 1 (URAT1)1 or glucose transporter 9 (GLUT9), respectively2 More renal uric acid transporters have been identified, both on the apical and basolateral sides of the proximal tubular cell membranes.3,4
Other than URAT1 and GLUT9, only ATP-binding cassette transporter G2 codified by the ABCG2 gene5 and sodium-dependent phosphate transporters type 1 and 4 encoded respectively by the SLC17A1 and SLC17A3 genes, have been related to human diseases but they present with hyperuricemia and gout and not hypouricemia.4,6 Nevertheless, there are reports of cases clinically diagnosed as RHUC without genetic resolution, suggesting that other transporters may be implicated diseases related with defects on renal UA handling.7
Incidence of RHUC has been estimated to be inferior to 1%, mostly from studies on the Asian population8,9 but populations from other geographic areas are also affected.10,11
Despite the efforts of estimating its prevalence, this condition is probably underdiagnosed, due to its clinical heterogeneity and lack of awareness from healthcare providers.
Although frequently asymptomatic12 RHUC may manifest with nephrolithiasis and nephrocalcinosis due to increased fractional excretion of uric acid (FEUA), and severe cases of exercise-induced acute kidney injury (EI-AKI).13,14
Data on long-term outcomes are lacking but due to the well-established relation between acute kidney injury (AKI) and chronic kidney disease (CKD), it seems likely that severe, repeated episodes of EI-AKI may lead to CKD and eventually end-stage kidney disease. We report the case of a man diagnosed with RHUC type 2 after hospitalization due to EI-AKI.
CASE REPORT
A 43-year-old man presented to the emergency department complaining of intense asthenia, right flank pain, nausea, and vomiting associated with a perception of decreased urinary output. The symptoms started some days after a 20 km walk during a hot day (outdoor temperature 30°C).
He had been diagnosed with type 2 diabetes mellitus, hypertension, and dyslipidemia 4 years before with no target organ damage diagnosed to that date. He was medicated with metformin 1 g bid, lisinopril/hydrochlorothiazide 20/12.5 mg id, fenofibrate 276 mg id and sinvastatin 40 mg id. He had no family history of kidney disease. His parents were born in close towns but there was no consanguinity.
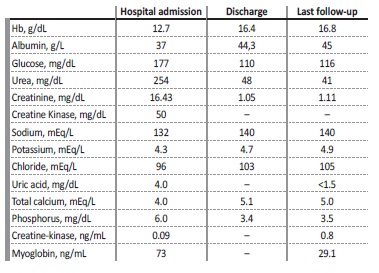
At hospital admission, he was described as being pale but vital signs were normal, with blood pressure 130/60 mmHg, heart rate 70 bpm, auricular temperature 36.5°C, and respiratory rate 14 cpm. Cardiac and pulmonary auscultation was unremarkable and there was no pedal edema. The abdomen had no alterations at sight, there was no tenderness at palpation, and Murphy’s renal sign was negative on both sides. Blood tests (Table 1) showed normal blood counts but severe azotemia, with serum creatinine (sCr) of 16.43 mg/dL, urea 254 mg/dL, glucose 118 mg/dL, UA 3.6 mg/dL, phosphorus 6.0 mg/dL, absence of analytic signs of rhabdomyolysis, mild hyponatremia 132 mEq/L and compensated metabolic acidosis with HCO3-of 17 mEq/L. Urinalysis had proteinuria 1+ but urinary sediment had no leukocyturia nor erythrocyturia. Glycosuria was not present. Proteinuria was of 700 mg on a 24-hour collection. Kidney ultrasound revealed normal-sized kidneys with preserved sinus-parenchymal differentiation and cortex thickness with no hydronephrosis, albeit some diffuse microcalculi. Doppler study showed no alterations in renal vasculature. The patient was admitted to the internal medicine ward and vigorous fluid therapy with normal saline was initiated alongside the withdrawal of ambulatory medication. The kidney function recovered rapidly, without requiring renal replacement therapy.
The patient was discharged after 15 days with a sCr of 1.66 mg/dL and complete resolution of the acid-base disorder, and the diagnosis of AKI in the context of dehydration was assumed. Three years after the initial episode he was referred for evaluation at the outpatient clinic for genetic kidney diseases. At that time, his blood tests disclosed a sCr of 1.05 mg/dL, urea of 48 mg/dL, UA below 1.5 mg/dL, and phosphorus of 3.4 mg/dL. Urinalysis presented glycosuria, assumed to be related to dapagliflozin, hyperuricosuria with FEUA above 32.5% (normal range: 5.5%-8.5%), and unremarkable urinary sediment. There was no history of stone emission nor evidence of a systemic disease affecting the tubular compartment. Therefore, the patient was proposed for genetic testing using Sanger sequencing for the genes of interest (SLC22A12 and SLC2A9). The molecular study identified the pathogenic variant c.1221del p.(His407Glnfs*8) at the SLC2A9 gene in homozygosity compatible with the diagnosis of RHUC type 2. To the best of our knowledge this mutation has not been described previously, and was not reported on Genome Aggregation Database (“gnomAD”). Afterwards, patient’s sister and mother, asymptomatic, were tested and both carried the same mutation, on heterozygosity. The father refused to be tested.The patient was advised to avoid intense and/or prolonged physical exercise, especially in hot environments, to ensure daily water ingestion over 3 L to prevent dehydration, and to avoid anti-hypertensive drugs with uricosuric effects, like losartan.
At the last follow-up, his blood pressure was well controlled with perindopril and amlodipine and he was also taking dapagliflozin, metformin, and linagliptin for glycemic control, as long as, fenofibrate and atorvastatin for dyslipidemia. There were no further records of EI-AKI episodes and his last sCr was 1.11 mg/dL.
DISCUSSION
We present a case of a patient diagnosed with RHUC type 2 due to the homozygous variant c.1221del p.(His407Glnfs*8) at SLC2A9 gene, detected 3 years after the development of a severe episode of EI-AKI. Despite being the most specific clinical manifestation, the diagnosis was not pursued at the presentation revealing the lack of awareness by the clinicians and consequently lack of the necessary therapeutic actions.
Serum UA levels rely on the balance between production and excretion. In normal conditions, two-thirds of urate are excreted in urine and the remaining one-third is eliminated by the intestine. Healthy individuals have FEUA between 5% and 10%.15 A shift in increasing UA production and/or decreasing excretion will lead to hyperuricemia and the opposite to hypouricemia. Both conditions have been associated with kidney disease.16
Based on current knowledge, either URAT1 or GLUT9 transporters dysfunction is involved in RHUC pathophysiology. URAT1 is located on the apical side of the proximal tubule cell membrane and is codified by the SLC22A12 gene17 and is a molecular target for some uricosuric drugs, such as losartan, irbesartan, and dotinurab. On the other hand, GLUT9 is located on the basolateral side of the proximal tubule cell membrane, and is codified by the SLC2A9 gene18 and is responsible for the uptake of urate to systemic circulation. Genome wide association studies has previously identified several loci implicated on sérum uric acid concentration on a population level, particularly SLC2A9 and ABG2.19 Furthermore, inactivating or loss-of-function mutations on SLC2A9 have been reported to cause significant hypouricemia.20
The most frequent clinical manifestation in RHUC is nephrolithiasis, easily justified by the highly saturated urine with UA. EI-AKI pathophysiology is still a matter of controversy. Some theories have been advocated. On one side, intratubular obstruction by UA could explain AKI during strenuous exercise, which is a well-known trigger of higher UA production which in association with volume depletion can promote its crystallization in renal tubules.21 Although this can be the culprit in some cases, most cases of EI-AKI related to RHUC show no deposition of UA crystals on renal biopsy specimens.22 On the other side, an enhanced renal vasoconstriction response in the presence of oxygen reactive species, not counteracted by UA, a potent antioxidant, has also been proposed.23 However, this hypothesis is counteracted by the absence of EI-AKI reports on patients with xanthinuria, who have also low UA levels due to absent production. A recently published experimental study reported a profound ATP loss in RHUC patients when compared with healthy and xanthinuric patients, proposing that the loss of hypoxanthine, a precursor of UA and a product of ATP/ AMP metabolism may cause ATP loss in the renal tubules with tissue damage.24 The development of a suitable mouse model may help to further clarify the mechanism underlying EI-AKI, with an investigational study using a double knockout for URAT1-Uricase mice model showing a temporary increase in UA serum levels and its urinary excretion during anoxic exercise leading to crystal formation and precipitation.
Furthermore, urine pH was decreased after exercise, contributing to kidney dysfunction.25 Finally, it has also been shown that EI-AKI in patients with RHUC, can be prevented by the administration of xanthine oxidoreductase inhibitors (XOI).25,26
To increase awareness of this disease and consequently improve diagnosis, a clinical practice guideline has been published concerning diagnosis criteria.27 They suggest that RHUC should be suspected based on a continuous serum UA level below 2 mg/dL and increased FEUA while excluding the various alternative clinical scenarios, such as Fanconi syndrome, Wilson’s disease, or syndrome of inappropriate secretion of antidiuretic hormone. As our case demonstrates and is also addressed in the guideline, during AKI episodes, serum UA level may be superior to the proposed threshold and should not lead to exclusion of RHUC. Genetic analysis of SLC2A9 and SLC22A12 demonstrating homozygous or compound heterozygous pathogenic mutations provides the definitive diagnosis of RHUC.
There is no specific treatment for this disease. Abundant water intake and avoidance of extenuating exercise are important to prevent the two main manifestations, nephrolithiasis and EI-AKI. The use of XOI protects patients from EI-AKI by decreasing UA production and its use is mandatory.25
Like the rest of the population, many of these patients will have common diseases such as diabetes or hypertension. A diagnosis of RHUC in this patient directly affects the drugs used to control these diseases.
Both losartan and irbesartan are uricosuric drugs by inhibiting the URAT1 transporter, and should not be used in this pathology, especially in RHUC type 2, where the URAT1 transporter is intact and probably determining some degree of UA reabsorption. Similarly, sodium-glucose transporter type 2 inhibitors, recommended on many guidelines as a first-line agente for diabetes treatment, should be prescribed with caution in these patients. These drugs lead to uricosuria in association with increased sodium excretion and glycosuria, leading to inhibition of urate proximal reabsorption28 and therefore promoting the formation of UA stones and increasing the likelihood of EI-AKI. In fact, enhancement of uric acid excretion by glycosuria seems to be dependent on URAT1 expression29 and therefore patients with SLC2A9 mutations may have a higher risk than patients with SLC22A12, if medicated with these drugs. This increased risk could be counteracted by increased water intake.
Regarding lesinurab, a recently developed drug used in the treatment of hyperuricemia acts by blocking URAT1, leading to increased excretion of UA, thus mimicking the RHUC type 1 phenotype. Data from phase 3 studies raised concern due to increased cardiovascular mortality and nephrotoxicity findings.30
In conclusion, we present a case of RHUC type 2 due to novel homozygous variant c.1221del p.(His407Glnfs*8) at the SLC2A9 gene diagnosed after an episode of EI-AKI, a typical presentation of this condition. Our case emphasizes the lack of awareness of this disease and its implications. Definite diagnosis with genetic test should be pursued since it leads to therapeutic and lifestyle changes. Although there is no specific treatment available, XOI such as allopurinol should be strongly considered.

