











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Waldenstrom’s macroglobulinemia (WM) is a B-cell lymphoproliferative disorder characterized by bone marrow infiltration by a lymphoplasmacytic lymphoma infiltration of bone marrow and a sérum immunoglobulin M (IgM) paraprotein.1,2 It is a rare lymphoid neoplasia, accounting for 2% of all hematological malignancies.3
WM can manifest with tissue infiltration by malignant cells, deposition of IgM and changes caused by specific properties of the sérum IgM, namely hyperviscosity. Although patients with WM can presente with an extensive range of signs and symptoms, most of them are related to tumoral infiltration (cytopenia and hepatosplenomegaly), monoclonal protein accumulation in the circulation (cryoglobulinemia and serum hyper viscosity syndrome), monoclonal protein accumulation in tissues (amyloidosis) or autoantibody production (neuropathy and hemolytic anemia).4,5
Although several pathogenic mechanisms that can affect multiple organs have been described regarding this disease, kidney involvement in WM is rare.2,6
The most common kidney biopsy findings include AL amyloidosis, cryoglobulinemic glomerulonephritis and direct infiltration of the kidney by atypical lymphoid cells along with large intracapillary aggregation of IgM. These injuries result from the infiltration of malignant cells, an immune mediated reaction to the cells or from the production of an abnormal paraprotein.2,7-9
We describe a case of WM with acute renal injury (AKI) and nephrotic syndrome (NS) due to a minimal change disease.
CASE REPORT
A 41-year-old Caucasian male presented to the hospital with a 3-week history of periorbital edema, slowly progressive bilateral lower extremity edema and weight gain. His past medical history includes MW diagnosis 16 years before. After diagnosis he received 8 cycles of R-CHOP (rituximab, cyclophosphamide, hydroxydaunorubicin hydrochloride, vincristine-oncovin and prednisolone) and after which his hematologist considered the patient to have an indolent form of MW, so the patient started a follow up with annual clinical and laboratorial evaluation. On physical examination the patient had a blood pressure of 146/96 mmHg, a 4+ bilateral lower extremity edema up to thighs, multiple palpable cervical, axillary, and inguinal lymph nodes and hepatosplenomegaly. The remaining clinical examination was unremarkable.
Laboratory results showed hypoalbuminemia (1.8 mg/dL), normal iron metabolism and beta 2 microglobulin was 10.786 mg/dL. Urinalysis showed of protein 400 mg/dL with a protein to creatinine ratio (P/C ratio) of 3.53 g/g. The remaining analytic results are presented in Table 1. Protein electrophoresis revealed a marked hypoalbuminemia and a monoclonal spike in the y region (Fig. 1 and Table 1). Immunoelectrophoresis showed an IgM kappa monoclonal protein (Table 1).
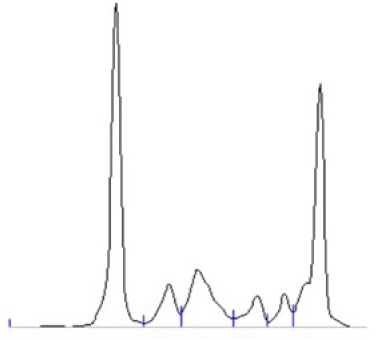
Table 1 Analytical results
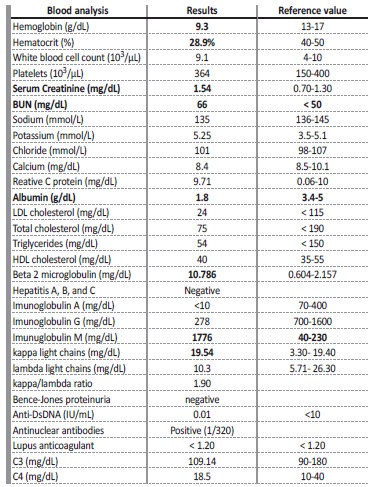
Anti-DsDNA - anti double stranded DNA; BUN - Blood urea nitrogen; HDL - High density lipoprotein
cholesterol; LDL-Low - density lipoprotein
Thorax, abdomen, and pelvis computed tomography revealed hepatosplenomegaly and multiple axillar lymphadenopathies, intra-abdominal and inguinal regions. The kidney ultrasound showed kidneys with preserved form and a slightly enlarged dimensions on the left; an obstructive component was excluded.
As a patient with a known history of WM, he was observed by ophthalmology and neurology and complications related to hyperviscosity were ruled out. The fat abdominal biopsy was negative for amyloidosis.
Kidney biopsy (left kidney) showed an interstitial lymphomatous infiltration (Fig. 2). Glomeruli were unremarkable. Immunohistochemistry showed interstitial staining for CD20 + B cells, (Fig. 3), CD5 - and CD10 -. Immunofluorescence was negative and electron microscopy showed the presence of diffuse effacement of podocytes foot processes and no electron dense deposits were presented. A diagnosis of minimal change disease (MCD) and renal parenchymal infiltration by lymphoplasmacytic lymphoma was made.
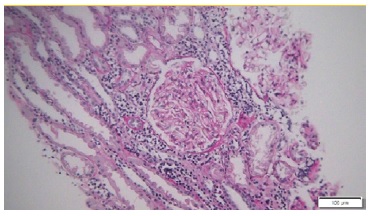
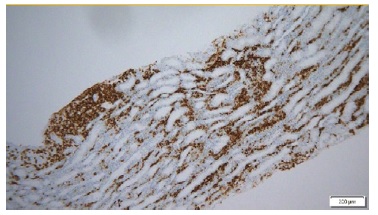
Figure 3 Immunohistochemistry for CD20: Lymphoplasmocytic infiltrate with CD 20 expression (ME 200x)
The patient was evaluated by hematology and steroids were started during hospitalization. After discharge, he was followed up in a hematology and nephrology outpatient clinic. He started treatment with bortezomib, dexamethasone and rituximab with progressive recovery of renal function (serum creatinine 1.01 mg/dL), and gradual remission of proteinuria after 6- months. (P/C ratio 0.185 g/g; serum albumin 3.8 g/dL).
DISCUSSION AND CONCLUSION
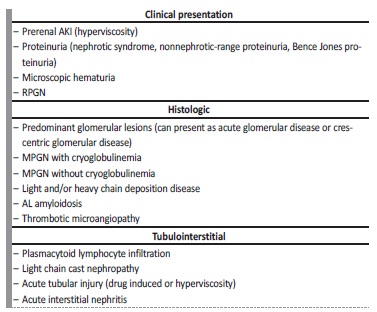
Although infrequent, kidney involvement in Waldenström macroglobulinemia is well documented (Table 2). Renal manifestations are most likely related to abnormal monoclonal protein and/or tissue infiltration by underlying lymphoplasmacytic neoplasm. Only less than 3% of the patient with WM develop end-stage renal disease and NS is present in less than 7% of the patients with WM.2,3,10,11
NS in the presence of lymphoplasmacytic lymphoma with IgM monoclonal gammopathy (WM) can have several causes but immunoglobulin light chain (AL) amyloidosis is the most frequent cause of NS in these patients, and non-amyloid nephrotic syndromes are very rare in the worldwide literature.6,7,12,13
Minimal change disease, a common complication of Hodgkin’s lymphoma, seems to occur very rarely in patients with WM.3,10,13
The pathophysiology of MCD remains poorly understood, however a T-lymphocyte disorder is suggested by the occurrence of the disease in the setting of immune challenge initiated by infectious diseases, drugs, allergies or vaccinations, and also the sensitivity to immunosuppressive agents. In WM, as well as in Hodgkin’s lymphoma patients, a T-cell disorder leading to a reduced CD4/CD8 ratio and an abnormal secretion of lymphokines has been documented and may be associated to the occurrence of MCD.12,14-18
AKI is also an uncommon presentation of MW. The most common causes of AKI in monoclonal gammopathies, such as hypercalcemia, uric acid nephropathy, dehydration, cast nephropathy, drug induced and sepsis, were not present in this case, so we admitted that the severe interstitial lymphocytic infiltrate was the cause of AKI.3
This case illustrates the importance of screening for renal disease in WM patients. Minimal change disease should be considered as a rare, but possible, cause of nephrotic syndrome in WM. A kidney biopsy should be performed in those presenting with otherwise unexplained AKI and/or nephrotic syndrome. Early detection of kidney disease and adequate treatment can preserve renal function while controlling the hematologic disease.19,20

