










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.20 no.4 Porto 2011
Um caso clínico de pseudohipoparatiroidismo
Helena Vieira1, Paula Vieira1
1 S. Pediatria, H São Francisco Xavier, CH Lisboa Ocidental
RESUMO
Introdução: O pseudohipoparatiroidismo engloba um grupo heterogéneo de doenças que se caracterizam por resistência dos órgãos alvo à acção da hormona paratiroideia.
Caso clínico: Descreve-se o caso clínico de uma criança de nove anos, com história de convulsões generalizadas e câimbras, de aparecimento aos quatro e seis anos, respectivamente. Apresentava dismorfia facial, cataratas, atraso na erupção das peças dentárias, sinais de Chvostek e Trousseau, atraso do desenvolvimento psicomotor e baixa estatura. Na tomografia cranio-encefálica evidenciavam-se calcificações intraparenquimatosas. A presença de hipocalcemia, hiperfosfatemia e aumento da hormona paratiroideia permitiu estabelecer o diagnóstico de pseudohipoparatiroidismo. O fenótipo de osteodistrofia hereditária de Albright associado a alterações da função tiroideia é sugestivo de pseudohipoparatiroidismo do tipo Ia, por ser o mais frequente. O doente manteve irregular adesão à terapêutica, o que motivou múltiplas descompensações metabólicas.
Conclusão: Salienta-se que a hipocalcemia deve ser considerada no diagnóstico diferencial das convulsões em idade pediátrica. O diagnóstico atempado e o correcto cumprimento da terapêutica, permitem prevenir as complicações da hipocalcemia.
Palavras-chave: fenótipo, hipocalcemia, pseudohipoparatiroidismo, convulsões.
A pseudohypoparathyroidism case report
ABSTRACT
Introduction: The pseudohypoparathyroidism includes a heterogeneous group of diseases characterized by parathyroid hormone action resistance of target organs.
Case report: We report the case of a nine year-old child with generalized convulsions and cramps since the age of four and six years, respectively. He presented dysmorphic features, cataracts, delayed teeth eruption, Chvostek and Trousseau signs, psychomotor delay and short stature. Brain tomographic scan revealed cerebral intraparenquimatous calcifications. The presence of hypocalcemia, hyperphosphatemia and increased parathyroid hormone established the diagnosis of pseudohypoparathyroidism. The phenotype of Albright hereditary osteodystrophy associated with changes in thyroid function are suggestive of pseudohypoparathyroidism Ia, the most frequent type. The patient had irregular adherence to therapy, which caused several metabolic decompensation.
Conclusion: We emphasize that hypocalcemia should be considered in the differential diagnosis of children seizures. Early diagnosis and proper compliance to therapy should prevent damage associated with hypocalcemia.
Keywords: phenotype, hypocalcemia, pseudohypoparathyroidism, seizures.
INTRODUÇÃO
Uma concentração constante de cálcio ionizado extracelular é essencial para a função celular, sendo mantida por um sistema homeostático. A etiologia da hipocalcemia pode estar relacionada com um dos componentes deste sistema, tal como alterações da hormona paratiroideia (PTH) ou da vitamina D, ou um defeito do receptor sensível ao cálcio. A PTH leva ao aumento do cálcio sérico através de três mecanismos: 1) aumento da reabsorção renal de cálcio; 2) aumento da reabsorção óssea; e 3) estimulação da 1-alfa-hidroxilase no rim, que converte a 25-hidroxivitamina D a 1,25-dihidroxivitamina D (a forma activa da vitamina D), a qual por sua vez aumenta a reabsorção renal e intestinal de cálcio. Para além da manutenção dos níveis plasmáticos de cálcio, a PTH promove o aumento da excreção urinária de fósforo e bicarbonato. (1-3)
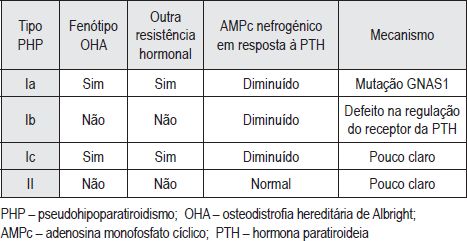
O pseudohipoparatiroidismo (PHP) engloba um grupo heterogéneo de doenças que se caracterizam por resistência dos órgãos alvo à acção da PTH, traduzindo-se por hipocalcemia, hiperfosfatemia, e concentrações plasmáticas elevadas de PTH. (2-6) Pode ser classificado em 4 tipos (Ia, Ib, Ic, II) de acordo com os achados fenotípicos, alterações bioquímicas e mecanismo genético (Quadro I).
Quadro I – Tipos de Pseudohipoparatiroidismo
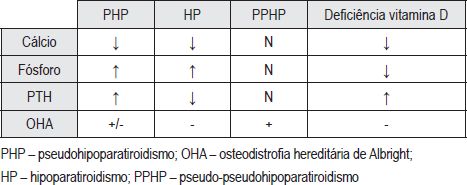
O hipoparatiroidismo, o pseudo-pseudohipoparatiroidismo (PPHP) e a deficiência em vitamina D, devem ser considerados no diagnóstico diferencial do PHP (Quadro II).
Quadro II – Diagnóstico diferencial de Pseudohipoparatiroidismo
Na descrição deste caso pretende-se alertar para os aspectos clínicos do pseudohipoparatiroidismo, e reforçar a importância da sua suspeição na abordagem das convulsões em idade pediátrica.
CASO CLÍNICO
Criança de nove anos, sexo masculino, natural de São Tomé e Príncipe. Sem antecedentes familiares patológicos de relevo. Sem informação da vigilância anterior no país de origem. Inicia aos quatro anos convulsões tonico-clónicas generalizadas, e aos seis anos câimbras nos membros superiores e inferiores. Aos oito anos são diagnosticadas cataratas bilaterais. Para esclarecimento do quadro clínico é transferido para internamento em Portugal, aos nove anos. Ao exame objectivo constatou-se a presença de face redonda e achatada, raiz nasal deprimida, cataratas bilaterais, atraso na erupção das peças dentárias, hipertonia muscular no tronco e membros superiores, sinais de Chvostek e Trousseau presentes, atraso do desenvolvimento psico-motor, baixa estatura (percentil 3) e estadio pubertário I de Tanner. Analiticamente destacava-se diminuição dos níveis séricos de cálcio total e de cálcio ionizado, aumento do fósforo, da fosfatase alcalina, e dos níveis de PTH (Quadro III). O cariotipo era normal. O electrocardiograma mostrou um prolongamento do intervalo QT. Na radiografia do esqueleto tinha redução da densidade óssea, sem outras alterações, sendo a idade óssea compatível com a idade cronológica. A tomografia cranio-encefálica (Figura 1) mostrou calcificações intraparenquimatosas dos núcleos lenticulares e caudados, tálamos, e substância branca fronto-parietal.
Quadro III – Avaliação laboratorial na admissão
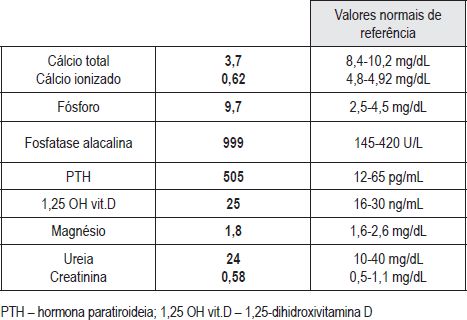
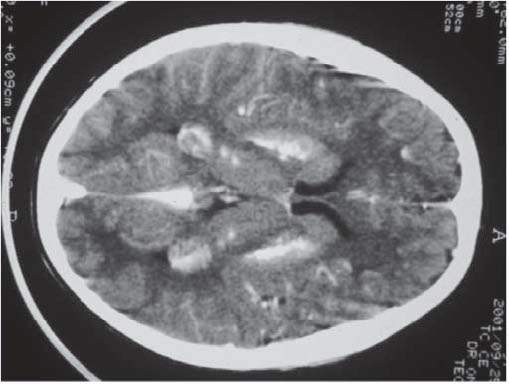
Figura 1 – Tomografia axial computorizada cranio-encefálica: calcificações intraparenquimatosas
Foi feita correcção da hipocalcemia com gluconato de cálcio endovenoso (e.v.), tendo mantido terapêutica em ambulatório com calcitriol e cálcio elementar. Manteve seguimento em consulta de Endocrinologia Pediátrica. A estatura evoluiu para o percentil 10 a 25. Foi submetido a cirurgia oftalmológica com correcção das cataratas aos onze anos. O cumprimento da terapêutica foi irregular, com controlo metabólico difícil e com vários internamentos por convulsões. Nas avaliações laboratoriais efectuadas regularmente manteve hipocalcemia, hiperfosfatemia e níveis elevados de PTH, com calciúria e 1,25-dihidroxivitamina D normais. A hormona estimuladora da tiróide (TSH) manteve valores normais ficando a tiroxina (T4) total diminuída. As gonadotrofinas mantiveram-se em níveis pré-puberes. Abandonou o seguimento em consulta ao fim de três anos.
DISCUSSÃO
Os primeiros casos de resistência à PTH foram descritos por Albright em 1942. Os doentes tinham hipocalcemia e hiperfosfatemia, e várias características fenotípicas, actualmente designadas de osteodistrofia hereditária de Albright (OHA).(3-5) Estas características incluem baixa estatura, face redonda, obesidade, hipoplasia do esmalte dentário, deformações das articulações (genu valgum, coxa vara, cubitus valgus), e alterações das mãos e pés, particularmente encurtamento dos ossos metacarpos e metatarsos do 4º e 5º dedos.(3-5,7,8) Entre 50 a 75% dos doentes têm atraso do desenvolvimento psicomotor ligeiro a moderado. A OHA normalmente não é reconhecida durante o primeiro ano de vida, tornando-se mais evidente aos 4-6 anos.(4) Nem todos os doentes com PHP têm o fenótipo OHA e por outro lado, o fenótipo de OHA está também presente em doentes com PPHP (Quadro II). O termo OHA é actualmente usado para descrever as características físicas, independentemente dos achados bioquímicos.(4,7)
O tipo mais comum de PHP é o Ia, uma doença autossómica dominante que tem características da OHA. Nestes casos a resistência à PTH resulta de uma mutação no gene GNAS1, o qual codifica a subunidade alfa da proteína G, acoplada ao receptor da PTH e necessária para a transdução de sinal nos tecidos alvo endócrinos.(2,4) A proteína G é assim incapaz de activar a adenilciclase para a produção de adenosina monofosfato cíclico (AMPc), necessária para a produção da resposta à PTH e manutenção da homeostase fosfo-cálcica.(2,3)
No caso descrito, a presença de hipocalcemia, hiperfosfatemia e aumento da PTH permitiu estabelecer o diagnóstico de pseudohipoparatiroidismo. Por estar associado a um fenótipo característico de OHA trata-se provavelmente do tipo Ia, pela sua maior frequência. No entanto para a sua confirmação teria sido necessário identificar a mutação no gene GNAS1.(2,6)
Muitos doentes com PHP Ia têm resistência a outras hormonas além da PTH, como a hormona libertadora da hormona de crescimento, glicagina, hormona adrenocorticotrófica, e mais frequentemente a TSH e as gonadotrofinas.(2,4,5-9) A resistência à TSH, clinicamente indistinguível do hipotiroidismo primário, pode tornar-se aparente antes que a hipocalcemia se desenvolva e, ocasionalmente, é diagnosticada através do rastreio metabólico neonatal.(9) O nível ligeiramente reduzido de T4 total (com TSH e T4 livre normais) neste doente, pode indiciar o desenvolvimento de hipotiroidismo. O hipogonadismo é mais comum nas raparigas e pode manifestar-se por atraso pubertário, alterações do ciclo menstrual, e infertilidade. Quando o defeito do gene GNAS1 é transmitido pela mãe, a resistência hormonal é expressa juntamente com o fenótipo OHA; se o defeito tem origem paterna, o fenótipo OHA ocorre isoladamente, já que o alelo materno normal resulta na manutenção da resposta à PTH.(2,4)
Por razões ainda desconhecidas, a maioria dos doentes que têm PHP tipo Ia tem concentrações normais de cálcio durante muitos anos, raramente demonstrando valores baixos de cálcio sérico antes dos três anos. Embora a tetania e o mal convulsivo sejam apresentações comuns da hipocalcemia em crianças em idade pré-escolar com PHP Ia, alguns doentes permanecem assintomáticos até à idade adulta. Radiologicamente pode haver evidência de calcificação progressiva dos gânglios cerebrais basais,(4,5) tal como ocorreu neste caso. Outra manifestação clínica do PHP tipo Ia são os nódulos subcutâneos.
O diagnóstico clínico pode ser confirmado pela demonstração de uma resposta diminuída no fósforo e AMPc urinários, após administração exógena de PTH. O diagnóstico definitivo é estabelecido pela identificação da mutação na proteína G.(2,6)
Os sintomas e sinais de hipocalcemia relacionam-se com a concentração de cálcio ionizado no soro.(2) A tetania, edema da papila, convulsões, prolongamento do intervalo QT no electrocardiograma, hipotensão e insuficiência cardíaca, ocorrem mais frequentemente em doentes que desenvolvem hipocalcemia agudamente.(3,7,10) Os achados clássicos que traduzem irritabilidade neuromuscular devido a tetania latente são os sinais de Chvostek e Trousseau.(3,10)
No entanto, as convulsões podem ser a única forma de apresentação. A presença de convulsões sem tetania, pode justificar-se pelo facto de que um nível baixo de cálcio ionizado no líquido cefalo-raquidiano poder ter um efeito convulsivo, mas não um efeito tetânico directo.(10) Pode-se assim explicar porque a apresentação inicial da hipocalcemia no nosso doente foram as convulsões.
A hipocalcemia crónica, entre outras características, traduz-se por alterações dentárias e da ectoderme, cataratas, calcificação dos gânglios basais, e baixa estatura, o que também se verificou neste caso clínico.(3,10)
Os doentes com hipocalcemia sintomática devem receber cálcio.(4,7) Os níveis séricos de fósforo normalizam frequentemente com a suplementação de cálcio e calcitriol, pelo que os quelantes do fósforo raramente são necessários. O seguimento regular é essencial para ajustar a terapêutica e optimizar o metabolismo fosfo-cálcico.(8)
CONCLUSÕES
O PHP é uma doença complexa com variabilidade individual. O diagnóstico é muitas vezes tardio, levando a uma abordagem e terapêutica iniciais inapropriadas.(8) É muito importante realizar uma investigação bioquímica completa do metabolismo fosfo-cálcico em todas as crianças que apresentem um quadro clínico sugestivo de hipocalcemia. O diagnóstico do PHP é habitualmente baseado em achados bioquímicos e fenotípicos. A caracterização molecular permite diferenciar os vários tipos de PHP; contudo não é imprescindível para o diagnóstico, nem para a adequada terapêutica e seguimento dos doentes.
O objectivo do tratamento é manter os níveis de cálcio ionizado dentro do intervalo normal, evitar a hipercalciúria, e normalizar os níveis de PTH.(3) Se a terapêutica for cumprida, podem prevenir-se as complicações agudas e crónicas da hipocalcemia e assim, reduzir a morbilidade.
BIBLIOGRAFIA
1. Zhou P, Markowitz M, Adam HM. Hypocalcemia in infants and children. Pediatr Rev 2009; 30:190-2. [ Links ]
2. Jeha G, Kirkland J. Etiology of hypocalcemia in infants and children. Janeiro, 2010. Acessível em: http://www.uptodate.com [ Links ]
3. Maeda S, Fortes E, Oliveira U, Borba V, Lazaretti-Castro M. Hypoparathyroidism and Pseudohypoparathyroidism. Arq Bras Endocrinol Metab 2006; 4: 664-73. [ Links ]
4. Burgert T, Markowitz M. Understanding and Recognizing Pseudohypoparathyroidism. Pediatr Rev 2005; 26: 308-9. [ Links ]
5. Kronenberg H. Williams Textbook of Endocrinology, 11ª ed. Philadelphia: Saunders Elsevier; 2008: 27. [ Links ]
6. Doyle D, DiGeorge A. Disorders of the Parathyroid. In: Behrman R, Kliegman R, Jenson H, Stanton B. Nelson Textbook of Pediatrics, 18ª ed. Saunders Elsevier; 2007: 2340-8. [ Links ]
7. Bujan M, Cervini A, Fano V, Pierini A. Osteodistrofia hereditaria de Albright: presentación de três casos clínicos. Arch Argent Pediatr 2010; 108: e24-e27. [ Links ]
8. Donghi V, Mora S, Zamproni I, Chiumello G, Weber G. Pseudohypoparathyroidism, an often delayed diagnosis: a case series. Cases J 2009; 2: 6734. [ Links ]
9. Balavoine A, Ladsous M, Velayoudom F, Vlaeminck V, Cardot-Bauters C, Herbomez M, et al. Hypothyroidism in patients with pseudohypoparathyroidism type Ia: clinical evidence of resistence to TSH and TRH. Eur J Endocrinol 2008; 159: 431-7. [ Links ]
10. Goltzman D. Clinical manifestations of hypocalcemia. Setembro, 2009. Acessível em: http://www.uptodate.com [ Links ]
Helena Vieira
E-mail: hrvieira@gmail.com

