










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.20 no.4 Porto 2011
Síndroma Cornelia de Lange e Disgenesia Cerebral
Ana Luísa Leite1, Marta Vila Real1, Fátima Santos1
1 S. Pediatria, CH Vila Nova de Gaia e Espinho
RESUMO
Introdução: A Síndroma Cornelia de Lange (SCdL) é uma síndroma polimalformativa rara, caracterizada por aparência facial peculiar, atraso de crescimento e do desenvolvimento psicomotor, alterações comportamentais e malformações major associadas (cardíacas, gastrointestinais e musculoesqueléticas).
Caso clínico: Os autores descrevem o caso duma lactente de seis meses, sexo feminino, com SCdL grave e disgenesia cerebral congénita em que a pesquisa de mutações no gene NIPBL foi negativa. Actualmente, aos três anos e meio, esta criança mantém seguimento por uma equipa multidisciplinar, com uma evolução clínica positiva, particularmente nas áreas motoras (postura e manipulação).
Discussão: O diagnóstico da SCdL é sobretudo clínico. A confirmação molecular pode ser útil em casos duvidosos, mas as mutações no gene NIPBL estão presentes em apenas 50% dos casos, pelo que perante um diagnóstico clínico seguro a ausência de mutações neste gene não exclui o diagnóstico. O diagnóstico precoce desta doença é útil para a programação de cuidados antecipatórios em relação às complicações mais frequentes e para aconselhamento genético aos pais.
Palavras-chave: Cornelia de Lange, disgenesia cerebral, síndroma.
Cornelia de lange syndrome and cerebral dysgenesis
ABSTRACT
Background: Cornelia de Lange Syndrome (CdLS) is a rare multiple malformation syndrome, characterized by specific facial features, small stature, developmental delay and major malformations (cardiac, gastrointestinal and musculoskeletal systems).
Case Report: The authors describe a clinical case of a six-month-old female toddler, with severe CdLS and congenital cerebral dysgenesis, in whom no NIPBL mutation was found. Nowadays, at the age of three and half years, this patient maintains a multidisciplinary approach and has a favourable evolution of her motor status (posture and manipulation).
Discussion: The diagnosis of CdLS is based in mostly on clinical grounds. Molecular confirmation can be helpful in atypical cases, but since mutations in the NIPBL gene are found in only around 50% of patients, failure to identify mutations does not exclude the diagnosis. An early diagnosis is important to program anticipatory surveillance for the most frequent complications of this condition and to provide adequate genetic counseling.
Keywords: Cornelia de Lange, cerebral dysgenesis, syndrome.
INTRODUÇÃO
A Síndroma Cornelia de Lange (SCdL) é uma síndroma polimalformativa caracterizada por aparência facial peculiar, atraso de crescimento, atraso do desenvolvimento psicomotor, alterações comportamentais e malformações major associadas (cardíacas, gastrointestinais e musculoesqueléticas).(1) Tem um espectro clínico vasto, variando desde fenótipos muito ligeiros até quadros graves e incompatíveis com a vida. (2)
Os autores apresentam um caso de SCdL clássico, com disgenesia cerebral congénita grave, dando especial relevância à metodologia diagnóstica e intervenção terapêutica com uma equipa multidisciplinar.
CASO CLÍNICO
Lactente do sexo feminino, com antecedentes de consanguinidade parental e hidrâmnios. Parto eutócico às 38 semanas, com Apgar 4/8 e necessidade de reanimação. Somatometria à nascença adequada à idade gestacional (peso 2920g, percentil 10; comprimento 49cm, percentil 25-50; perímetro cefálico 34cm, percentil 25).
Internada no primeiro dia de vida por episódio de cianose com a mamada; efectuou ecocardiograma, por apresentar sopro cardíaco, que foi normal. Durante os primeiros meses de vida manteve frequentes e abundantes episódios de regurgitação gastroesofágica, apesar das medidas anti-refluxo instituídas.
Aos seis meses foi orientada para consulta de Neuropediatria por apresentar atraso de desenvolvimento psicomotor (ADPM), má evolução estaturo-ponderal e microcefalia com plagiocefalia.
Apresentava face dismórfica (Figura 1), alterações das extremidades (camptodactilia do 4º e 5º dedos das mãos, polegares de implantação proximal, braquidactilia dos dedos dos pés), e pele marmoreada com mamilos e cicatriz umbilical hipoplásicos. O choro era atípico (agudo, de intensidade baixa, tipo ronronar) e apresentava ADPM grave com movimentos oculares erráticos, hipotonia axial marcada, sem controlo cefálico, postura em batráquio e movimentos discinéticos dos membros superiores.

Figura 1 – Face dismórfica: sobrancelhas arqueadas, sinófrio, pestanas longas, ponte nasal deprimida, base do nariz larga, filtro longo e apagado, lábios finos, implantação baixa dos pavilhões auriculares e do cabelo, micrognatia, hipertrofia gengival.
A RM cerebral evidenciou uma malformação cerebral, com disgenesia grave (Figura 2).
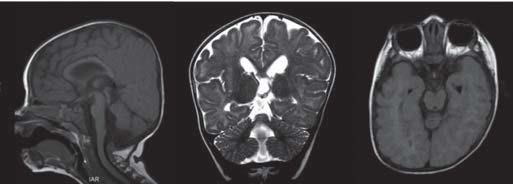
Figura 2 – Atrofia do tronco, sobretudo da protuberância e pedúnculos cerebelosos médios, sendo o IV ventrículo proeminente; discreto alargamento dos sulcos da convexidade vermiana superior; proeminência do sistema ventricular supratentorial, IV ventrículo e espaços de líquor da convexidade.
Perante as alterações clínicas concluiu-se tratar de uma síndrome polimalformativa, compatível com o diagnóstico de síndroma de Cornelia de Lange do tipo I (clássico), com evidência de disgenesia cerebral congénita grave.
O cariótipo foi normal (46,XX). A pesquisa de mutações no gene NIPBL foi negativa. A ecografia reno-vesical foi normal. A radiografia esofagogastroduodenal contrastada revelou pequena aspiração de contraste para a árvore traqueobrônquica, excluindo existência de hérnia do hiato.
Inicialmente não foram constatadas alterações oftalmológicas significativas, contudo, com o crescimento revelou estrabismo e miopia (compensada com lentes correctivas). A avaliação audiológica revelou hipoacúsia bilateral grave, no entanto, sem tolerância para o uso de próteses auditivas. A sua evolução estaturo-ponderal mantém-se abaixo do percentil 5 para a idade e sexo, contudo, a doença de refluxo gastroesofágico (DRGE) tem sido bem controlada com associação de omeprazol e domperidona.
Actualmente, aos três anos e meio, é acompanhada por uma equipa multidisciplinar (Neuropediatra, Geneticista, Gastrenterologista, Oftalmologista, Ortopedista, Otorrinolaringologista, Nutricionista e Fisioterapeutas/Terapeutas Ocupacionais e Professor de Ensino Especial) com uma evolução clínica positiva particularmente nas áreas motoras (postura e manipulação).
Numa gravidez seguinte, este casal teve um segundo filho com síndroma polimalformativa grave, caracterizado por: dismorfia facial, agenesia dos pavilhões auriculares, estenose das coanas, cardiopatia (comunicação inter-ventricular e estenose mitral, com hipertensão pulmonar), epilepsia e hipotonia generalizada. Este filho faleceu nos primeiros meses de vida, no decurso de complicação após cirurgia cardíaca, não tendo a etiologia desta síndroma sido completamente esclarecida.
DISCUSSÃO
O SCdL é uma síndrome genética rara, com incidência estimada de cerca de 1:10.000 casos. Contabilizando as formas ligeiras admite-se uma real incidência superior. Não apresenta predilecção racial e é ligeiramente mais frequente no sexo feminino (F/M:1,3/1). A etiologia é ainda incerta em muitos doentes e a maioria dos casos são esporádicos. O risco de recorrência estimado é de 2-5% para irmãos de um indivíduo afectado, filho de pais saudáveis, dada a possibilidade de mosaicismo gonadal num dos progenitores; quando um dos progenitores é afectado o risco de recorrência aumenta para 50%.(1)
O diagnóstico é fundamentalmente clínico, tendo sido estabelecidos critérios diagnósticos mínimos, após o consenso entre a Fundação Americana de Cornelia de Lange e o Comité Científico Mundial para o SCdL.(1) Os critérios clínicos devem incluir: a aparência facial e três ou mais características das descritas abaixo (Tabela 1), bem como o preencher dois ou três critérios dentro dos seis sistemas afectados (pelo menos um deve pertencer a uma das três áreas major: crescimento, desenvolvimento e comportamento).
Tabela 1 – Critérios Diagnósticos para SCdL (1).
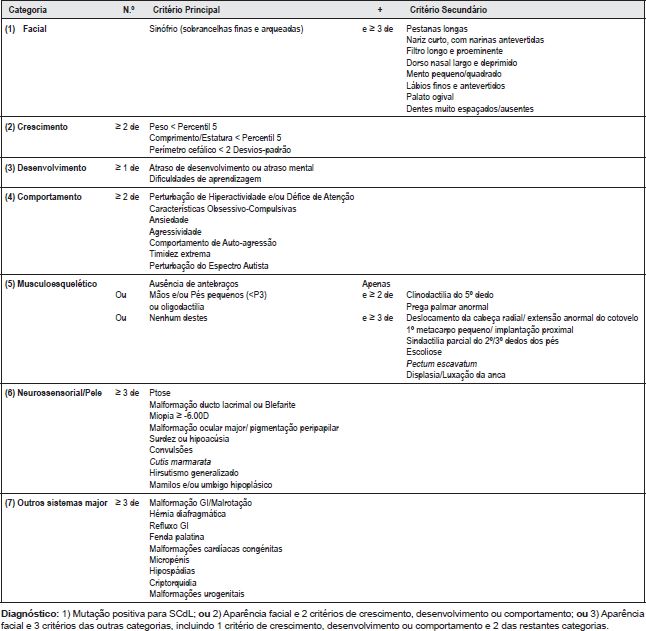
No caso clínico apresentado cumprem-se os parâmetros diagnósticos clínicos faciais, de crescimento e desenvolvimento, músculo-esqueléticos, cutâneos e gastrointestinais (GI).
Em 1993, Van Allen et al propôs um sistema de classificação para a SCdL em 3 tipos. No Tipo I (Clássico) os doentes exibem as alterações faciais e esqueléticas características desde a nascença. No Tipo II (Ligeiro) exibem características semelhantes às do tipo I em algum momento da vida, tornando-se mais evidentes após os 2-3 anos de vida e associadas a ADPM ligeiro, na ausência de malformações major. No Tipo III (fenocópia de SCL) englobam doentes com fenótipo compatível com SCdL devido a alterações cromossómicas ou exposição teratogénica. (2)
As lesões cerebrais nem sempre estão presentes no SCdL. Contudo, a sua presença contribui inevitavelmente para o atraso de desenvolvimento e alterações neurológicas típicas destes doentes. As lesões podem ser do tipo hipóxico-isquémicas ou alterações por disgenesia congénita cerebral. As lesões do tipo hipóxico-isquémico são as mais frequentes e geralmente ocorrem periparto, secundárias a patologia cardíaca cianótica congénita. As alterações por disgenesia cerebral, pelo contrário, são mais raras e condicionam essencialmente microcefalia, imaturidade/ simplicidade das circunvoluções dos giros cerebrais e alterações ao nível do diencéfalo e sistema cortico-ponto-cerebeloso, não estando ainda bem definido o momento em que afectam a morfogénese. (3-5)
Kline et al, estabeleceu em 1996 um sistema de avaliação da gravidade, que correlaciona alterações cerebrais específicas com o nível de desenvolvimento. Sendo assim, os doentes mais graves são aqueles com atrofia dos lobos frontais ou atrofia global (com alargamento dos ventrículos), agenesia do vermis cerebeloso inferior, alargamento da cisterna magna, dos cornos frontais e dos sulcos na RMN cerebral, como no nosso caso clínico. (6)
Recentemente, foram descritas alterações genéticas associadas ao SCdL. As mutações do gene NIPBL (braço curto do cromossoma 5) foram descritas pela primeira vez em 2004 por Kratz et al e actualmente pensa-se que seja o gene mais vezes associada ao fenótipo clássico. (7) Na Europa estima-se que metade dos doentes com SCdL apresente esta mutação. (8) As mutações nos genes SMC1L1 (cromossoma X) e SMC3 (cromossoma 10q), estão mais vezes associadas a formas ligeiras. (9)
O diagnóstico pré-natal (na ausência de caso índex) mantém-se um desafio, sendo possível conjugar alterações ecográficas características (ex. aumento da translucência da nuca, RCIU simétrico, anomalias dos membros/face) com o estudo molecular do gene NIPBL em ADN fetal (10,11), sendo que a ausência de mutações, mais uma vez, não permitirá excluir o diagnóstico. Martinez-Frias et al descrevem a idade jovem dos pais como um possível factor de risco para SCdL.(12) Em famílias com mutação identificada previamente o diagnóstico pré-natal é possível através da pesquisa dessa mutação em ADN fetal.
No nosso caso clínico, houve uma gestação posterior também consanguínea, não programada e mal vigiada, que cursou com o nascimento de um indivíduo masculino com várias malformações. Clinicamente destacaram-se a cardiopatia major, agenesia dos pavilhões auriculares, estenose das coanas e um fácies dismórfico. Contudo, pelo desfecho fatal precoce, e na ausência da identificação de mutações no gene NIPBL, o diagnóstico clínico de SCdL neste doente não é seguro. Esta evolução negativa reforça ainda mais a necessidade de haver um acompanhamento rigoroso da grávida durante a gestação de forma a permitir um diagnóstico malformativo atempado e um adequado aconselhamento genético.
Os doentes com SCdL, pelo envolvimento multissistémico, têm necessidade de acompanhamento multidisciplinar o qual com frequência envolve uma equipa composta por médicos, nutricionistas e terapeutas.(1) De acordo com a gravidade clínica e as áreas mais atingidas poderá ser necessária maior abrangência de sub-especialidades. O pediatra é fundamental na coordenação entre as várias sub-especialidades e terapêuticas, nomeadamente a estimulação precoce para optimizar o seu desenvolvimento psicomotor.
CONCLUSÃO
A existência de lesão cerebral na Síndroma Cornelia de Lange, tal como a disgenesia cerebral, é determinante no prognóstico destas crianças.
O pediatra tem um papel importante no diagnóstico precoce e na orientação dos doentes com Síndroma Cornelia de Lange. O seguimento por uma equipa multidisciplinar é importante para a programação de cuidados em relação às complicações mais frequentes e ao aconselhamenteo genético.
BIBLIOGRAFIA
1. Kline AD, Krantz ID, Sommer A, Kliewer M, Jackson LG, FitzPatrick DR, et al. Cornelia de Lange Syndrome: Clinical Review, Diagnostic and Scoring Systems and Antecepatory Guidance. Am J Med Genet (Part A) 2007; 143A:1287-96. [ Links ]
2. Van Allen MI, Filippi G, Siegel-Bartelt J, Yong SL, Ritchie S, Toi A, Reynolds JF. Clinical variability within Brachmann-de-Lange syndrome: a proposed classification system. Am J Med Genet 1993, 47:947-58. [ Links ]
3. Yamaguchi K, Ishitobi F. Brain dysgenesis in Cornelia de Lange syndrome. Clin Neuropathol 1999; 18:99-105. [ Links ]
4. Vuilleumier N, Kövari E, Michon A, Hof P, Mentenopoulos G, Giannakopoulos P, Bouras C. Neuropathological analysis of an adult case of the Cornelia de Lange syndrome. Acta Neuropathol 2002; 104:327-32. [ Links ]
5. Hayashi M, Sakamoto K, Kurata K, Nagata J, Satoh J, Morimatsu Y. Septo-optic dysplasia with cerebellar hypoplasia in Cornelia de Lange syndrome. Acta Neuropathol 1996;92:625-630. [ Links ]
6. Kline AD, Jackson LG, Kliewer M. A scoring system for clinical severity correlates with brain findings in Cornelia de Lange syndrome. Am J Hum Genet 69:280. [ Links ]
7. Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 2004; 36:631-5. [ Links ]
8. Bhuiyan ZA, Klein M, Hammond P, VanHaeringen A, Mannens MMAM, VanBerckelaer-Onnes I, Hennekam RCM. Genotype-phenotype correlations of 39 patients with Cornelia De Lange syndrome: the Dutch experience. J Med Genet 2006; 40:568-75. [ Links ]
9. Musio A, Selicorni A, Focarelli ML, Gervsini C, Milani D, Russo S, et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet 2006; 38: 528-30. [ Links ]
10. Urban M, Hartung J. Ultrasonographic and clinical appearance of a 22-week-old fetus with Brachmann-de Lange syndrome. Am J Med Genet 2001; 102:73-5. [ Links ]
11. Lalatta F, Russo S, Gentilin B, Boschetto C, Cavalleri F, Masciadri M, et al. Prenatal/neonatal pathology in two cases of Cornelia de Lange syndrome harboring novel mutations of NIPBL. Genet Med 2007; 9:188-94. [ Links ]
12. Martínez-Frías ML, Bermejo E, Jiménez N, Gómez-Ullate J, López JA, Aparicio P, et al. Brachmann-de-Lange síndrome in our population: clinical and epidemiological characteristics. An Esp Pediatr 1998; 48:293-8. [ Links ]
Ana Luísa Leite
E-mail: ana.luisa20@gmail.com

