










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.20 no.4 Porto 2011
Caso electroencefalográfico
Tânia Monteiro1, Esmeralda Martins2, Rui Chorão3
1 S. Neurologia Pediátrica, H Maria Pia, CH Porto
2 U. Metabolismo, S. Pediatria, H Maria Pia, CH Porto
3 U. Neurofisiologia Pediátrica, H Maria Pia, CH Porto
ABSTRACT
Introduction: The suppression-burst (SB) electroencephalographic pattern is rather common during the neonatal period and suggests severe encephalopathy. When significant hypoxic-ischemic insult is excluded, brain malformations and metabolic disorders have to be ruled out. Two distinctive epileptic syndromes are described: early epileptic encephalopathy with SB (Ohtahara syndrome) and early myoclonic epilepsy (EME). The later is frequently associated with neurometabolic disorders, one of the most common being nonketotic hyperglycinemia (NKH).
Case report: A baby girl presented with multiple erratic clonic and myoclonic seizures from the second day of life, refractory to antiepileptic drugs. She was hypotonic, lethargic and had episodes of apnea. The electroencephalogram (EEG) showed multiple bursts of multifocal epileptiform activity with long periods of almost flat tracing; this pattern persisted beyond the neonatal period, it was present at the last EEG performed at age four months. Barbiturate-induced coma with mechanical ventilation was induced. She died at the age of five months.
The second but not the first sample of cerebrospinal fluid (CSF) and blood revealed an increased CSF/serum glycine ratio (0,11 – normal<0,03). Post-morten liver tissue biopsy found a deficit at the glycine cleavage system (GCS) (6,6 mkat/ kg - normal 45,0-195,0) and molecular studies detected a mutation in the gene GLDC molecular testing. This result allowed better parents genetic counseling.
Conclusions: Early myoclonic epilepsy presents with multifocal seizures and SB on pattern on the EEG in the neonatal period, metabolic causes must be investigated, namely the neonatal form of NHK. CSF and plasma aminoacids, including glycine levels, should be measured, simultaneously and sometimes repeatedly. Enzymatic and molecular analysis may confirm this diagnosis and are useful for parents genetic counseling.
Keywords: Supression-burst EEG pattern, early myoclonic epilepsy, nonketotic hyperglycinemia
Apresenta-se o caso de lactente do sexo feminino, primeira filha de pais saudáveis não consanguíneos, parto eutócico às 36 semanas, Índice de Apgar 8/9/10, sem necessidade de manobras de reanimação e somatometria adequada. Sem história familiar de doença neurológica.
No segundo dia de vida apresentava hipotonia e letargia e iniciou convulsões associadas a episódios de apneia. No quinto dia de vida, por agravamento progressivo, necessitou de suporte ventilatório, com transferência para uma Unidade de Cuidados Intensivos Pediátricos. À entrada apresentava crises mioclónicas e clónicas multifocais, hipotonia com flutuações do tónus, letargia, face inexpressiva e ausência de contacto visual. Realizou vídeo-EEG, que mostrava actividade epileptiforme multifocal com padrão de surto-supressão (Figuras 1 e 2). Os períodos de supressão eram assíncronos e com duração até 10 segundos ou mais. Registaram-se crises clínicas, clónicas, e crises eléctricas focais com localização variável. Este padrão persistiu para além do período neonatal (Figuras 3 e 4).
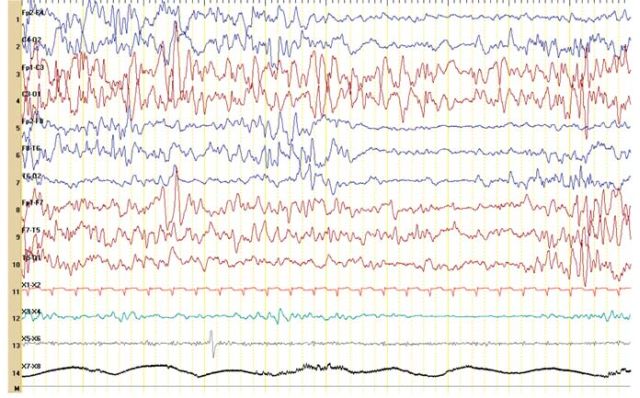
Figura 1 – Actividade paroxística multifocal (idade corrigida: 38 semanas 4 dias).
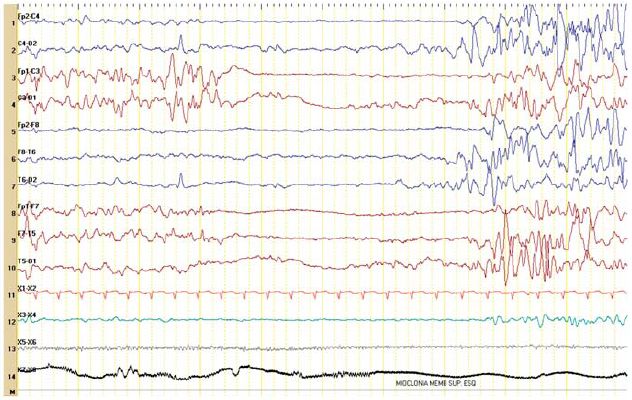
Figura 2 – Padrão de surto-supressão (idade corrigida: 38 semanas 4 dias).
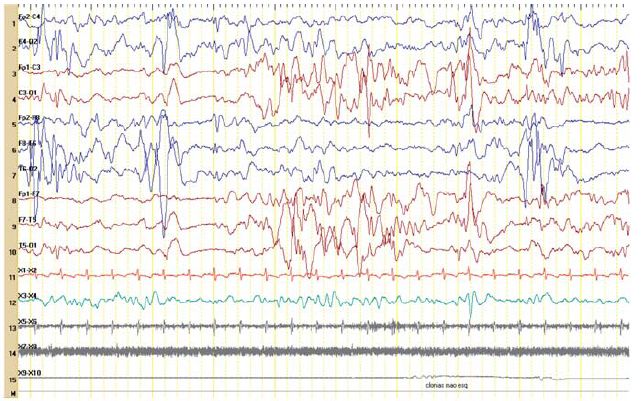
Figura 3 – Actividade epileptiforme multifocal e registo de crises clónicas focais (idade: 4 meses).
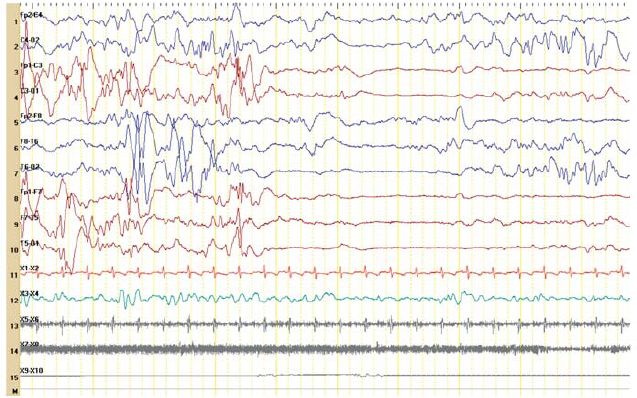
Figura 4 – Persistência de actividade paroxística multifocal com trechos de supressão (4 meses de idade).
Fez ressonância magnética cerebral com um mês de vida, normal; repetiu aos cinco meses (atrofia difusa). Não foi realizada espectroscopia.
Manteve crises epilépticas refractárias à múltipla terapêutica: fenobarbital, fenitoína, valproato de sódio, topiramato e pirodixina. Necessitou de indução de coma com tiopental, mantendo ventilação mecânica com traqueostomia ao mês e meio de vida. Faleceu aos cinco meses de vida.
Qual o diagnóstico clínico-electroencefalográfico?
Qual a investigação complementar?
DIAGNÓSTICO
Epilepsia neonatal grave com padrão de surto-supressão (epilepsia mioclónica precoce)
Perante este diagnóstico é essencial excluir erros inatos do metabolismo, sendo uma das causas metabólicas mais frequentes a hiperglicinemia não cetótica (HGNC) ou encefalopatia glicínica.
Nesta doente a relação glicina no líquido cefalorraquidiano (LCR) / plasma com um mês de vida foi normal (0,03), assim como o estudo dos restantes aminoácidos no plasma e LCR. A segunda amostra, colhida aos três meses de idade, mostrou uma relação aumentada (0,11, com valor de referência <0,02).
No tecido hepático post-morten foi detectado défice no sistema de clivagem da glicina (6,6 μkat/ kg - normal 45,0 -195,0) e o estudo molecular com presença de uma mutação no gene GLDC, o que confirmou o diagnóstico de hiperglicinemia não cetótica (HGNC) de apresentação neonatal.
DISCUSSÃO
O padrão electroencefalográfico de surto-supressão, caracterizado por uma sucessão de surtos de actividade paroxística intercalados por episódios de traçado de muito baixa amplitude ou plano é relativamente comum no período neonatal e frequentemente associado a convulsões. Ocorre em casos de lesões anoxico-isquémicas, mas nestas é geralmente transitório, ao contrário do que acontece quando associado a malformações cerebrais ou doenças metabólicas.(1)
O síndrome de Ohtahara, também conhecido por encefalopatia epiléptica infantil precoce, é uma entidade rara, quase sempre associada a lesões estruturais graves. As crises mioclónicas são raras, sendo frequentes os espasmos tónicos e crises focais. Na maioria dos casos o prognóstico é mau, com epilepsia refractária.(1)
A epilepsia mioclónica precoce, como no caso apresentado, manifesta-se com mioclonias erráticas e crises focais motoras. Há casos criptogénicos e até familiares, mas importa reter que se associa muitas vezes a doenças neurometabólicas – mais frequentemente HGNC, mas também a acidúrias orgânicas (D-glicérica, metilmalónica, isovalérica e propiónica). (1,4)
Na forma neonatal de HGNC, além do padrão de crises epilépticas, são característicos a hipotonia, letargia e apneia de causa central. (2-4)
A hiperglicinemia não cetótica é uma doença de transmissão autossómica recessiva em que ocorre um defeito no sistema de clivagem da glicina (SCG) que leva à sua acumulação nos tecidos corporais, nomeadamente no cérebro, sendo responsável pela maioria dos sintomas, principalmente no período neonatal. (2-4) O SCG é um complexo de quatro proteínas (P,H,T,L), localizado no interior da membrana mitocondrial do fígado, rim, cérebro e placenta. A maioria das mutações ocorrem no gene da descarboxilase da glicina GLDC (locus 9p22, codifica a P-proteína, 70-75% dos casos). Estão descritas também mutações no gene AMT (locus 3p21.2-p21.1, codifica a T-proteína, cerca de 20%) e GCSH (locus 16q23.2, codifica a H-proteína, em menos de 1%).(2)
O diagnóstico faz-se pelo aumento de glicina na urina, plasma e líquor, na ausência de uma acidúria orgânica, com aumento da relação glicina líquor/plasma (3,4) (diagnóstico se >0,08; se >0,04 requer pesquisa do défice enzimático do SCG (biópsia de fígado ou em linfoblastos) ou estudo molecular. Este, se bem que muitas vezes não necessário para o diagnóstico, interessa sobretudo para aconselhamento genético. (4)
Existem outras duas formas desta doença; uma rara, transitória, com o mesmo fenótipo inicial da neonatal e uma forma tardia.(4)
As alternativas terapêuticas disponíveis são de eficácia limitada: a dieta hipoproteica; o benzoato de sódio, que promove a diminuição da glicina através da eliminação renal, principalmente a nível plasmático; antagonistas do receptor do NMDA, como o dextrometorfano (actualmente não comercializado), que inibe o efeito excitotóxico da glicina no cérebro, sendo um potente antiepiléptico em alguns casos, e a quetamina que parece melhorar de forma parcial os sintomas neurológicos e a necessidade de suporte ventilatório. As benzodiazepinas (diazepam) são consideradas mais eficazes que outros antiepilépticos, devido ao efeito competidor dos receptores de glicina. (3,4) Os outros antiepilépticos são ineficazes; em particular, o valproato está contra-indicado na HGNC, por aumentar a concentração de glicina no líquor e, assim, as complicações neurológicas. (2)
O prognóstico destes doentes é, excepto nas formas transitórias, muito reservado, com mortalidade significativa no primeiro ano de vida e sequelas neurológicas importantes nos sobreviventes.(4)
CONCLUSÃO
O padrão electroclínico da epilepsia mioclónica precoce leva a suspeitar de doença metabólica, sendo a hiperglicinemia não cetótica uma das mais comuns. A investigação deve ser exaustiva nestes casos, de forma permitir o tratamento adequado precoce e o aconselhamento genético.
BIBLIOGRAFIA
1. Aicardi J, Ohtahara S. Severe neonatal epilepsies with suppression-burst pattern. In: Roger J, Bureau M, Dravet Ch, Genton P, Tassinari CA, Wolf P. Epileptic syndromes in infancy, childhood and adolescence. 4th ed. Montrouge: John Libbey Eurotext; 2005. p. 39-50. [ Links ]
2. Dhamija R, Mack, JK. A 2-day-old baby girl with encephalopathy and burst suppression on EEG. Neurology 2011;77: e16-9. [ Links ]
3. Neonatal non-ketotic hyperglycinemia: Report of five patients. Pediatr Int 2008;50: 121-3. [ Links ]
4. Hoover-Fong JE, Shah S, Van Hove JL, Applegarth D, Toone J, Hamosh A. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology;63: 1847-53. [ Links ]

