










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.21 no.1 Porto 2012
Síndrome de Miller Fisher numa criança
Dora Gomes1, Filipa Leite1, Nuno Andrade1, Mónica Vasconcelos2, Conceição Robalo2, Isabel Fineza2
1 S. Pediatria, H São Teotónio, Viseu
2 Centro de Desenvolvimento da Criança Luís Borges, Consulta de Neuropediatria, H Pediátrico de Coimbra
RESUMO
Introdução: O síndrome de Miller Fisher, variante do síndrome de Guillain-Barré, é uma doença desmielinizante inflamatória aguda, que é rara em idade pediátrica. O seu diagnóstico é baseado na tríade oftalmoplegia, ataxia e arreflexia. Em cerca de metade dos casos está descrita uma intercorrência infecciosa precedendo os sintomas neurológicos em cinco a dez dias.
Caso clínico: Os autores relatam o caso de uma criança de cinco anos de idade com disartria, ataxia e oftalmoplegia após episódio de gastroenterite aguda na semana prévia ao início da sintomatologia. À observação apresentava disartria, parésia bilateral do VI par, fraqueza muscular distal (de predomínio nos membros direitos) com ausência dos reflexos osteotendinosos aquilianos. A investigação analítica e imagiológica inicial não revelou alterações. O resultado do electromiografia foi compatível com poliradiculoneuropatia subaguda. O diagnóstico de síndrome Miller Fisher foi efectuado após exclusão de outras etiologias. A evolução clínica foi favorável, sem insuficiência respiratória ou outras complicações, com melhoria gradual dos défices neurológicos. Houve recuperação da ataxia ao fim de quatro semanas e da oftalmoplegia três meses após o diagnóstico.
Conclusões: O síndrome Miller Fisher é extremamente raro em idade pediátrica e constitui um desafio diagnóstico neste grupo etário. O prognóstico é habitualmente favorável. A propósito deste caso são discutidos os principais diagnósticos diferenciais.
Palavras-chave: síndrome de Miller Fisher, síndrome de Guillain-Barré, doença desmielinizante, criança.
A child with miller fisher syndrome
ABSTRACT
Background: Miller Fisher syndrome, a variant of Guillain-Barré syndrome, is an acute inflammatory demyelinating disease that is rare in children. The diagnosis is based on the triad of ophthalmoplegia, ataxia and areflexia. In about half of the cases there is an infectious complication preceding neurologic symptoms in five to ten days.
Case report: We describe the case of a five year-old boy who presented with a three-day history of diplopia, dysarthria and gait disturbance following an acute gastroenteritis. On examination he was found to have ataxia, areflexia and ophthalmoplegia. The laboratorial and imaging investigations were normal. The results of electromyogram were consistent with subacute polyradiculoneuropathy. The diagnosis of Miller Fisher syndrome was made after the exclusion of other conditions. The clinical outcome was favorable without respiratory failure or other complications, with gradual improvement of neurological deficits. Ataxia was restored in four weeks and ophthalmoplegia improved three months later.
Conclusions: Miller Fisher syndrome is extremely rare in children and is a diagnostic challenge at those ages. Outcome is usually good. This report outlines the frequency of Miller Fisher syndrome and lists the differential diagnoses that should be considered.
Keywords: Miller Fisher syndrome, Guillain-Barré syndrome, demyelinating disease, child.
INTRODUÇÃO
O síndrome de Guillain-Barré (SGB) é a causa mais comum de paralisia flácida aguda nas crianças. Trata-se de uma polineuropatia periférica de início agudo, de natureza auto-imune, que é precedida por um episódio infeccioso em mais de metade dos casos. Classicamente caracteriza-se por paralisia progressiva e simétrica, geralmente com carácter ascendente, e arreflexia. A paralisia dos músculos respiratórios com necessidade de ventilação mecânica é uma complicação possível do SGB. Pode ocorrer atingimento dos nervos cranianos. Estão habitualmente associadas alterações disautonómicas (arritmias, hipotensão postural, flutuações da tensão arterial, sinais vasomotores)(1). O diagnóstico é essencialmente clínico. O estudo do líquido cefalorraquidiano (LCR) e a electromiografia (EMG) são importantes adjuvantes diagnósticos. A análise do LCR mostra aumento da proteinorraquia e contagem celular <10 células/mm3 (dissociação albumino-citológica). A electrofisiologia revela diminuição das velocidades de condução nervosa sensitiva e motora, compatível com desmielinização. O prognóstico é geralmente favorável. A recuperação tem início duas a quatro semanas após paragem da progressão e na criança geralmente esta é completa. As sequelas neurológicas estão descritas em 5 a 25% dos doentes e a recorrência em 3% dos casos.
Em 1932, Collier descreveu a tríade ataxia, arreflexia e oftalmoplegia como uma variante do síndrome Guillain-Barré(2). No entanto, em 1956, Miller Fisher relatou três doentes com esta tríade clínica, mas como uma entidade separada, mais tarde denominada Síndrome Miller Fisher (SMF)(2,3). Este quadro neurológico é, actualmente, considerado uma doença desmielinizante inflamatória aguda do sistema nervoso periférico, afectando principalmente os nervos cranianos(3,4). Desde que este síndrome foi descrito pela primeira vez, em 1956, até 1992 foram relatados 223 casos, destes apenas 32 em crianças. A incidência estimada na criança é de 2-8 casos/10 milhões(2,4). É mais frequente no sexo masculino, com uma razão de 2:1, com um pico de incidência nos meses da Primavera(2,5,6). Em dois terços dos casos está descrita uma intercorrência infecciosa, geralmente infecção das vias aéreas superiores ou gastroenterite aguda, precedendo os sintomas neurológicos em cinco a dez dias(2,7,8). Os microrganismos mais frequentemente descritos têm sido o Haemophilus influenzae e Campylobacter jejuni. A presença de anticorpos anti-IgG GQ1b está associada ao SMF(4,9). Em 60% dos doentes observa-se hiperproteinorraquia.
O curso natural do SMF é caracterizado por uma boa recuperação, com pouca ou nenhuma incapacidade funcional 10 semanas, em média, após o início da sintomatologia(2,5).
Apresentamos o caso clínico de uma criança de cinco anos de idade com o objectivo de acrescentar mais informação a uma entidade neurológica rara em Pediatria, visto que existem, até à data, poucas revisões casuísticas de SMF publicadas. A propósito deste caso clínico são discutidos os principais diagnósticos diferenciais.
CASO CLÍNICO
Os autores relatam o caso de uma criança de cinco anos de idade, do sexo masculino, que, sete dias após episódio de gastroenterite aguda, inicia diplopia, alteração da voz e desequilíbrio. Este quadro teve início com diminuição da força muscular dos membros direitos a que se associou limitação da mobilidade ocular e disfonia. Teria ingerido mel na véspera do início da sintomatologia.
Recorreu ao serviço de urgência do Hospital local (hospital nível II) com o quadro descrito com três dias de evolução. Ao exame objectivo foi constatada disartria, parésia bilateral do VI par, fraqueza muscular distal de predomínio nos membros direitos e ausência de reflexos osteotendinosos aquilianos.
As hipóteses de diagnóstico colocadas inicialmente foram: síndrome Miller Fisher, botulismo e acidente vascular cerebral. A investigação inicial, que incluiu glicemia, hemograma e bioquímica sérica, pesquisa de tóxicos na urina, pesquisa de toxina botulínica nas fezes e tomografia computorizada cerebral, não revelou quaisquer alterações.
Por agravamento clínico com recusa na marcha e prostração foi transferido para um hospital nível III para apoio de Neuropediatria e complemento de neuroimagem colocando-se nessa altura as hipóteses de diagnóstico de encefalomielite aguda disseminada (ADEM), síndrome Miller Fisher e botulismo(4,10).
Realizou ressonância magnética (RM) cerebral e medular que não evidenciou alterações. O exame do LCR, realizado ao quarto dia de doença, não revelou aumento das proteínas nem pleocitose. O índice de IgG e as bandas oligoclonais do LCR foram normais. A pesquisa de anticorpos anti-GQ1b não pôde ser realizada. Não foram efectuadas coproculturas. Realizou EMG com estudo de condução nervosa, uma semana após o início dos sintomas, que mostrou uma diminuição das velocidades de condução nervosa compatível com polirradiculoneuropatia subaguda. Este resultado permitiu o diagnóstico de síndrome de Miller Fisher.
Ao sexto dia de internamento, constatou-se agravamento da disartria e ataxia, com aparecimento de disfagia. No entanto assistiu-se a melhoria clínica 48 horas depois, de tal modo que a criança teve alta ao 11º dia. Nunca houve compromisso respiratório durante todo o internamento.
A evolução clínica desta criança foi favorável, com resolução completa da ataxia quatro semanas após o início da sintomatologia e melhoria progressiva da oftalmoplegia aos três meses.
DISCUSSÃO
O síndrome Miller Fisher é raro na criança, o que torna o seu diagnóstico difícil. Pode estar associado a doenças infecciosas, autoimunes e neoplásicas. No caso apresentado há referência a gastroenterite aguda na semana prévia ao início do quadro.
O diagnóstico de síndrome Miller Fisher é essencialmente clínico, pelo que na sua suspeita há que excluir outras causas de fraqueza muscular, ataxia ou oftalmoplegia. Neste caso, o diagnóstico de SMF foi sugerido atendendo essencialmente à clínica e efectuado após exclusão de outras etiologias. Realça-se que na criança há com frequência formas atípicas de apresentação, o que implica excluir entidades que cursam com alteração da vigília como a encefalite e encefalomielite aguda disseminada, que se apresentam com ataxia como tumores cerebrais e intoxicações, que surgem com défice neurológicos focais como doença vascular cerebral e esclerose múltipla e ainda entidades que se manifestam com oftalmoplegia como a miastenia gravis e o botulismo. Assim sendo o diagnóstico diferencial deve ser ser feito com as neuropatias periféricas (tóxicas e infecciosas), a poliomielite (sobretudo a vacinal), mielopatia aguda por compressão medular (traumatismo, tumor, abcesso), doença vascular cerebral, doença dismielinizante do sistema nervoso central e doença da placa neuromuscular (botulismo, miastenia gravis) (Quadro 1).
Quadro 1 Diagnóstico diferencial de síndrome Miller Fisher
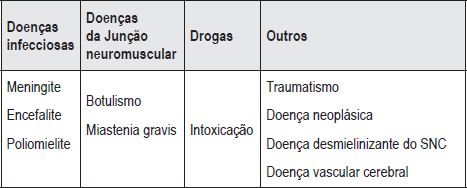
Os exames complementares, nomeadamente o exame do LCR e EMG podem ser normais no início do quadro, pelo que é fundamental um alto índice de suspeição. No nosso caso, não se verificou dissociação albumino-citológica no LCR o que pode ser explicado pela precocidade da realização da punção lombar dado que a elevação da proteinorraquia ocorre tardiamente (após a primeira semana)(10). Nos casos em que a punção lombar é realizada na primeira semana de doença, pode ser necessário repetir posteriormente para esclarecimento diagnóstico. A EMG é útil para a confirmação do diagnóstico, sobretudo na presença de características atípicas, mas também pode ser normal na primeira semana(10). Neste caso clínico o resultado da EMG veio corroborar o diagnóstico de síndrome Miller Fisher. A RM cerebral e medular permitiu excluir doença do sistema nervoso central nomeadamente doença desmielinizante.Na investigação etiológica poderia ter interesse neste caso a realização de coprocultura com pesquisa de Campylobacter jejuni. Não foi efectuada a pesquisa de anticorpos anti-gangliosídeo GQ1b, o que poderia ter reforçado o diagnóstico. A patogenia do SMF é atribuída a mecanismo auto-imune contra os antigénios dos nervos periféricos. Em cerca de 90% dos doentes com SMF podemos encontrar anticorpos anti-gangliosídeo GQ1b. Está descrita também reactividade cruzada destes anticorpos com epítopos de Campylobacter jejuni. Os gangliosídeos GQ1b constituem um componente lipídico abundante dos nervos oculomotores o que explica a oftalmoplegia(11,12).
A abolição dos reflexos osteotendinosos sugere o atingimento nervoso periférico, achado quase constante nesta entidade. A principal diferença entre SMF e as variantes mais comuns do SGB é que os grupos nervosos a serem afectados em primeiro lugar nos doentes com SMF são os dos pares cranianos, resultando em alteração dos movimentos oculares e desequilíbrio. A paralisia em outras formas de SGB é, na maioria dos casos, geralmente ascendente com fraqueza muscular progressiva com início nos membros inferiores.
Atendendo à boa evolução clínica do caso apresentado, sem perda total da autonomia da marcha e sem necessidade de ventilação mecânica, não se efectuou tratamento com imunoglobulina endovenosa. O tratamento com imunoglobulina ou plasmaferese no SMF está reservado para as formas clínicas moderadas e graves. Considera-se o uso de imunoglobulina endovenosa ou plasmaferese na doença rapidamente progressiva, com incapacidade de caminhar sem ajuda, agravamento da função respiratória e necessidade de ventilação mecânica e na paralisia bulbar significativa(10). No entanto, não há estudos randomizados realizados em idade pediátrica e os estudos de evidência de imunoglobulina no SMF são escassos(13,14). O uso de corticoterapia não está indicado no SMF por falta de evidência científica(15).
O SMF é habitualmente uma situação auto-limitada, como aconteceu no caso clínico descrito. O prognóstico é quase sempre favorável, com uma boa recuperação e sem défices residuais. No entanto, existem casos descritos com evolução para insuficiência respiratória e necessidade de ventilação mecânica. A recuperação habitualmente começa dentro de duas a quatro semanas após o início dos sintomas e está completa dentro de seis meses(2).
CONCLUSÕES
O síndrome Miller Fisher é uma entidade neurológica rara em idade pediátrica e constitui um desafio diagnóstico neste grupo etário. A tríade clínica característica (ataxia, arreflexia e oftalmoplegia) e os exames de imagem, nomeadamente a RM cerebral, exame do LCR e a EMG ajudam a confirmar o diagnóstico e a excluir outras situações que podem ser clinicamente sobreponíveis. A terapêutica é de suporte, sendo necessária vigilância e monitorização contínua destes doentes. A imunoglobulina endovenosa ou plasmaferese estão indicadas na doença moderada ou grave. O prognóstico é habitualmente favorável, com recuperação total que ocorre em média ao fim de seis meses.
BIBLIOGRAFIA
1. Hahn FA. Guillain-Barré syndrome. Lancet 1998; 352: 635-41. [ Links ]
2. Berlit P, Rakicky J. The Miller Fisher syndrome, review of the literature. J Clin Neuroophtalmol 1992; 12: 57-63. [ Links ]
3. Fisher M. An unusual variant of the acute idiopathic polyneuritis (Syndrome of ophtalmoplegia, ataxia and areflexia). N Engl J Med 1956; 255: 57-65. [ Links ]
4. Garrett J, Ryan P. A child with Miller Fisher syndrome. J Paediatr Child Health 2002; 38: 413-6. [ Links ]
5. Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher syndrome. Neurology 2001; 56: 1104-6. [ Links ]
6. Lo YL. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle Nerve 2007; 36: 615-27. [ Links ]
7. Bushra J. Miller Fisher Syndrome: An Uncommon Acute Neuropathy. The Journal Emergency Medicine 2000; 18: 427-30. [ Links ]
8. Overell JR, Willison HJ. Recent developments in Miller Fisher syndrome and related disorders. Curr Opin Neurol 2005; 18:562-6. [ Links ]
9. Benito-Leon J, Bravo J, Mateos F, Simon R. Miller Fisher syndrome in infancy. Child Nerv Syst 1996; 12: 559-61. [ Links ]
10. Sampaio MJ, Figueiroa S, Temudo T, Gomes S, Janeiro P, Silva R. Síndrome de Guillain-Barré – Protocolo de actuação (Julho de 2010). Sociedade Portuguesa de Neuropediatria. [ Links ]
11. Sánchez Torrent L, Noguera Julian A, Pérez Dueñas B, Nascimento Osorio A, Colomer Oferil J. Síndrome de Miller Fisher en la edad pediátrica: descripción de 3 casos. An Pediatr (Barc) 2009; 71: 377-8. [ Links ]
12. Kusunoki S, Chiba A, Kanazawa I. Anti-GQ1b IgG antibody is associated with ataxia as well as ophthalmoplegia. Musce Nerve 1999; 22: 1071-4. [ Links ]
13. Overell JR, Hsieh ST, Odaka M, Yuki N, Willison HJ. Treatment for Fisher syndrome, Bickerstaffs brainstem encephalitis and related disorders. Cochrane Database Syst Rev 2007;24: CD004761. [ Links ]
14. Donofrio PD, Berger A, Brannagan TH, Bromberg MB, Howard JF, Latov N, et al. Consensus statement: The use of intravenous immunoglobulin in the treatment of neuromuscular conditions report of the AANEM ad hoc committee. Muscle Nerve 2009; 40: 890-900. [ Links ]
15. Hughes RAC, Swan AV, van Koningsveld R, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010; 17: CD001446. [ Links ]
Dora Gomes
E-mail: doragomes@gmail.com

