










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.21 no.1 Porto 2012
Paraplegia espástica familiar tipo 4 – antecipação ou variabilidade fenotípica?
Nádia Rodrigues1, Sofia Ferreira1, Lia Rodrigues1, Ana Castro1, Célia Barbosa2, Roseli Gomes2
1 S. Pediatria, H Pedro Hispano, ULS Matosinhos
2 U. Neuropediatria, H Pedro Hispano, ULS Matosinhos
RESUMO
Introdução: A Paraplegia Espástica Familiar (PEF) é um grupo heterogéneo de doenças neurodegenerativas hereditárias, com uma prevalência de 2/100000 indivíduos na população portuguesa. Caracteriza-se sobretudo por espasticidade progressiva e insidiosa dos membros inferiores, por degeneração do feixe corticoespinhal.
Caso Clínico: Apresentamos uma criança com espasticidade e hiperreflexia progressivas dos membros inferiores, com vários familiares da linha paterna com sintomatologia semelhante, sugerindo ao diagnóstico de paraplegia espástica familiar. Foi identificada no probando uma mutação causal de paraplegia espástica familiar tipo 4.
Conclusão: Nesta família, a idade de início variou entre os cinco e os 50 anos, e diminuiu em média 22,5 anos ao longo de três gerações e, por outro lado, a apresentação e evolução da doença foram aparentemente mais graves em gerações sucessivas, sugerindo a existência de fenómeno de antecipação.
Palavras-chave: doença de Strümpell-Lorrain, paraplegia espástica familiar, paraplegia espástica familiar tipo 4, espastina, antecipação, variabilidade fenotípica.
Hereditary spastic paraplegia type 4 – anticipation or phenotypic variability?
ABSTRACT
Introduction: Hereditary spastic paraplegia is a heterogeneous group of inherited neurodegenerative diseases, with a prevalence of 2/100000 in the Portuguese population. It is mainly characterized by progressive and insidious spasticity of the lower limbs due to degeneration of corticospinal tracts.
Case Report: We present a child with progressive spasticity and hyperreflexia of lower limbs, with several relatives of the paternal line with similar symptoms, suggesting the diagnosis of hereditary spastic paraplegia. A causing mutation of hereditary spastic paraplegia type 4 was identified in the proband.
Conclusion: In this family, the age of onset varies from five to 50 years, and decreased in average 22,5 years over three generations. The clinical presentation and progression apparently tended to be more severe in successive generations, witch these suggests the phenomenon of anticipation.
Keywords: Strümpell-Lorrain disease, hereditary spastic paraplegia, hereditary spastic paraplegia type 4, spastin, anticipation, phenotypic variability.
INTRODUÇÃO
A Paraplegia Espástica Familiar (PEF), também conhecida como Doença de Strümpell-Lorrain, é um grupo heterogéneo de doenças hereditárias neurodegenerativas, sendo reportadas prevalências díspares na literatura variando de 0,5 a 12/100000(1-4). Em Portugal, Silva et al, estima uma prevalência de 2/100000(5). São características comuns às várias formas da doença a fraqueza muscular e espasticidade progressivas e insidiosas dos membros inferiores. A penetrância é incompleta e dependente da idade, é estimada em 85% aos 45 anos de idade(1).
Fisiopatologicamente caracteriza-se por degenerescência das terminações do feixe corticoespinhal, com atingimento predominante das terminações das fibras mais longas que suprem os membros inferiores.
A maioria são de transmissão autossómica dominante, e cerca de 40% destas formas correspondem ao tipo 4(5-7). A paraplegia espástica familiar tipo 4 está associada à mutação do gene que codifica a espastina (SPG4). Apresenta elevada variabilidade inter e intrafamiliar quer na idade de início quer na evolução clínica(1,7,8). Alguns autores descrevem o fenómeno de antecipação para alguns tipos da doença, nomeadamente no tipo 4, ou seja, doença de início mais precoce e mais grave em gerações subsequentes(1,9-11).
O diagnóstico é sugerido fundamentalmente pela clínica sugestiva e história familiar. Existe diagnóstico molecular em várias formas da doença, possibilitando o diagnóstico pré-natal e diagnóstico genético pré-implantatório.
CASO CLÍNICO
Criança do sexo feminino, cinco anos de idade, referenciada à consulta de Neurologia Pediátrica por dificuldades na marcha com quedas frequentes, cansaço fácil e tendência a marcha em bicos de pés. Os antecedentes pré ou peri-natais e neonatais eram irrelevantes. Ao nível do desenvolvimento psico-motor era notório o atraso na aquisição das etapas motoras, com início da marcha com apoio aos 18 meses e sem apoio aos 24 meses de idade e sempre com grandes dificuldades na corrida, sendo o desenvolvimento cognitivo normal. Na história familiar eram evidentes sintomas semelhantes em vários familiares da linha paterna, nomeadamente o pai de 33 anos, com antecedentes de cansaço fácil com a marcha desde os 25 anos e actualmente com grandes dificuldades em deambular e com disartria e disfagia; o tio paterno de 28 anos com sintomas idênticos desde os 25 anos e já com grandes dificuldades de deambulação; a avó paterna de 53 anos que desde os 50 anos está depende de apoio de cadeira de rodas; e a bisavó paterna de 72 anos com dificuldades em deambular sem apoio, desconhecendo a idade do início dos sintomas (Figura 1).
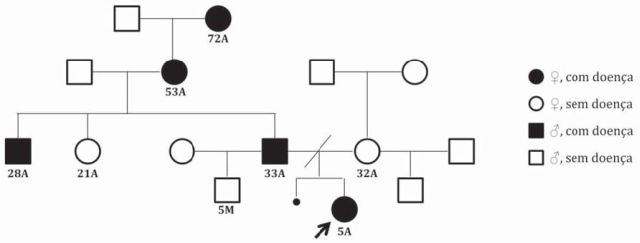
Figura 1 Heredograma
Ao exame neurológico apresentava dificuldades na marcha em calcanhares, sem qualquer limitação na marcha em bicos de pés, hiperreflexia limitada aos membros inferiores, clónus esgotável dos tornozelos, Babinski bilateral e limitação da dorsiflexão das articulações tibiotársicas. A força muscular e os reflexos osteotendinosos dos membros superiores eram normais. As sensibilidades (dolorosa, térmica, vibratória e proprioceptiva) estavam preservadas. O restante exame objectivo era normal.
Perante a clínica de fraqueza, espasticidade e hiperreflexia dos membros inferiores numa criança com vários familiares com sintomatologia semelhante foi colocada como hipótese diagnóstica a PEF. A apresentação familiar sugeria uma forma da doença de transmissão autossómica dominante.
A ressonância magnética cerebral e medular foi normal, nomeadamente, sem alterações de sinal da substância branca e em particular dos feixes piramidais, da estrutura ou morfologia do corpo caloso.
O estudo molecular foi dirigido inicialmente à pesquisa de mutações, delecções e duplicações do gene SPG3A, por ser a forma autossómica dominante que frequentemente se manifesta na infância. Contudo, o estudo por DHPLC (denaturing high-performance liquid chromatography) não identificou mutações, nem a análise por MLPA (multiplex ligation-dependent probe amplification) identificou delecções ou duplicações do gene SPG3A. Dada a forte suspeita clínica de estarmos perante uma forma autossómica dominante da doença, foi ainda realizado o estudo molecular do gene SPG4, por DHPLC, que identificou a mutação missense c.1158T>G (p.Asn386Lys) em heterozigotia no exão 8, mutação já descrita por outros autores e considerada causal(12), confirmando o diagnóstico de PEF tipo 4.
DISCUSSÃO
A heterogeneidade clínica e genotípica são uma característica chave da PEF. Mesmo dentro de cada família não existe consistência fenotípica, sendo manifesta a grande variabilidade na idade de início dos sintomas, na gravidade clínica e evolução da doença. Apesar de na PEF tipo 4, a idade média de início dos sintomas serem os 30 anos, a doença poder-se-á apresentar em qualquer idade. A doença tipicamente evolui inexoravelmente, habitualmente de forma insidiosa, apesar de alguns indivíduos permanecerem apenas ligeiramente afectados ou mesmo assintomáticos(7). Esta variabilidade justifica a necessidade de uma vigilância adequada de familiares assintomáticos.
No presente caso é evidente que em gerações sucessivas a doença se manifestou em idades progressivamente mais precoces e os sintomas são sucessivamente mais graves e rapidamente progressivos, podendo este facto traduzir o fenómeno de antecipação ou simplesmente a conhecida variabilidade fenotípica da doença. Nesta família, a idade de início varia dos cinco aos 50 anos e diminui em média 22,5 anos ao longo das três gerações.
Os cinco indivíduos descritos nesta família distribuídos por três gerações, não constituem número suficiente para concluir ou excluir antecipação.
O fenómeno de antecipação, ou seja, a diminuição da idade de início dos sintomas e/ou o aumento da gravidade dos sintomas em geração subsequentes tem sido descrito em estudos que envolvem várias famílias afectadas pela doença(11) ou que estudam apenas uma família mas com muitos indivíduos afectados pela PEF(10). Outros autores, pelo contrário, não encontram evidências suficientemente consistentes que confirmem este fenómeno na PEF tipo 4(7).
Apesar de a maioria dos indivíduos afectados por esta forma da doença apresentarem a forma pura da doença, a forma complicada da doença tem sido descrita por vários autores(1,13). As formas complicadas associam-se a outras manifestações, como défice cognitivo, epilepsia, retinopatia e neuropatia óptica, ataxia, disartria, surdez, neuropatia periférica e ictiose, entre outras. Nesta família, o pai do probando apresenta sintomas rapidamente progressivos e também sinais de disfunção bulbar como a disartria e disfagia, o que sugere uma forma complicada, não manifesta nos restantes familiares.
Deverá suspeitar-se de PEF em indivíduos com clínica característica e história familiar positiva. Deverá ponderar-se o estudo genético molecular após a exclusão de causas estruturais de paraplegia espástica. Dada a heterogeneidade genotípica, o estudo molecular deverá ser dirigido pela clínica e forma de hereditariedade subjacente. O estudo molecular do gene SPG4 deverá ser realizado em doentes com a forma pura de paraplegia espástica familiar autossómica dominante de início na idade adulta e o estudo do gene SPG3A em formas puras autossómicas dominantes de início na infância. Contudo, à semelhança do descrito neste caso clínico, em crianças afectadas em que há familiares com doença de início na idade adulta, deve solicitar-se o estudo molecular do gene SGP4, quer pela sua frequência quer pelo fenómeno de antecipação, que tem vindo a ser associado a esta forma de PEF.
A disponibilidade de confirmar, em várias formas da doença, o diagnóstico clínico com o estudo genético molecular, permite oferecer a possibilidade de diagnóstico pré-natal e diagnóstico genético pré-implantatório, quando a mutação, delecção ou duplicação causais são conhecidas.
CONCLUSÃO
O diagnóstico de paraplegia espástica familiar deve ser considerado em doentes com sintomatologia característica, nomeadamente fraqueza muscular e espasticidade limitadas aos membros inferiores, de evolução progressiva e insidiosa, e história familiar positiva. O diagnóstico molecular deverá ser dirigido pela clínica e formas de hereditariedade subjacentes a cada caso individualmente. São, contudo, necessários mais estudos que envolvam grande número de famílias, para esclarecer se o fenómeno de antecipação está de facto associado à PEF tipo 4 ou se se trata apenas de variabilidade fenotípica.
BIBLIOGRAFIA
1. Dürr A, Tallaksen C, Depienne C. Spastic Paraplegia Type 4. Genereviews. 2009. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1160/ [ Links ]
2. Erichsen AK, Koht J, Stray-Pedersen A, Abdelnoor M, Tallaksen CM. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain 2009; 132: 1577-88. [ Links ]
3. Braschinsky M, Luus SM, Gross-Paju K, Haldre S. The prevalence of hereditary spastic paraplegia and the occurrence of SPG4 mutations in Estonia. Neuroepidemiology 2009; 32: 89-93. [ Links ]
4. McMonagle P, Webb S, Hutchinson M. The prevalence of pure autosomal dominant hereditary spastic paraparesis in the island of Ireland. J Neurol Neurosurg Psychiatry 2002; 72: 43-6. [ Links ]
5. Silva MC, Coutinho P, Pinheiro CD, Neves JM, Serrano P. Hereditary ataxias and spastic paraplegias: methodological aspects of a prevalence study in Portugal. J Clin Epidemiol 1997; 50: 1377-84. [ Links ]
6. McDermott CJ, Burness CE, Kirby J, Cox LE, Rao DG, Hewarnadduma C, et al. Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology 2006; 67: 45-51. [ Links ]
7. Dürr A, Davoine CS, Paternotte C, von Fellenberg J, Cogilinicean S, Coutinho P, et al. Phenotype of autosomal dominant spastic paraplegia linked to chromosome 2. Brain 1996; 119: 1487-96. [ Links ]
8. Santorelli FM, Patrono C, Fortini D, Tessa A, Comanducci G, Bertini E, et al. Intrafamilial variability in hereditary spastic paraplegia associated with an SPG4 gene mutation. Neurology 2000; 55: 702-5. [ Links ]
9. Reddy PL, Seltzer WK, Grewal RP. Possible anticipation in hereditary spastic paraplegia type 4 (SPG4). Can J Neurol Sci 2007; 34: 208-10. [ Links ]
10. Thurmon TF, He C, Haskell C, Thorpe P, Thurmon SG, Rosen DR. Genetic anticipation in a large family with pure autosomal dominant hereditary spastic paraplegia.Am J Med Genet 1999; 83: 392-6. [ Links ]
11. Raskind WH, Pericak-Vance MA, Lennon F, Wolff J, Lipe HP, Bird TD. Familial spastic paraparesis: evaluation of locus heterogeneity, anticipation, and haplotype mapping of the SPG4 locus on the short arm of chromosome 2. Am J Med Genet. 1997; 74: 26-36. [ Links ]
12. Fonknechten N, Manel D, Byrne P, Davoine CS, Cruaud C, Bönsch D, et al. Spectrum of SGP4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet 2000; 9: 637-44. [ Links ]
13. Rowland LP, Bird TD. Silver syndrome: The complexity of complicated hereditary spastic paraplegia. Neurology 2008; 70:1948-9. [ Links ]

