










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.21 no.1 Porto 2012
Genes, crianças e pediatras
Esmeralda Martins1, Teresa Oliveira1, Anabela Bandeira1
1 U. Doenças Metabólicas, H Maria Pia, CH Porto
ABSTRACT
Homocystinuria is an autosomal recessive disease due to cystathionine-synthase deficiency, with the gene CBS being located in chromosome 21. In its typical presentation the eye, skeleton, central nervous system, and vascular system are all involved. The patient is normal at birth and in non-treated patients tall stature and ectopia lentis may be the first symptoms, as in the case we present.
CASO CLÍNICO
Segunda filha de um casal não consanguíneo sem história de doenças heredo familiares. Gestação vigiada sem intercorrências, parto eutócico hospitalar às 39 semanas. A somatometria ao nascimento foi: peso 3100 gr (P 50), comprimento 49 cm (P 50) e perímetro cefálico 34 cm (P 50). O período neonatal decorreu sem intercorrências.
Entre os um e quatro meses de idade o crescimento é rápido com passagem do percentil do comprimento de P 50 para P 90 como se pode ver na Figura 1, passando para um P superior a 95 após o ano de idade. O peso mantém-se no P50 e o perímetro cefálico no P 75.
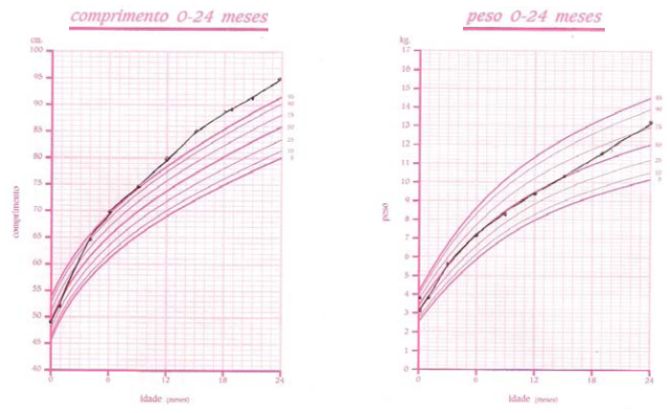
Figura 1 - Crescimento estaturoponderal
O desenvolvimento psicomotor era normal para a idade. Após iniciar a escolaridade, aos seis anos de idade, é notada diminuição da acuidade visual tendo sido encaminhada para observação por oftalmologia onde foi detetada uma luxação bilateral e inferior do cristalino (Figura 2).
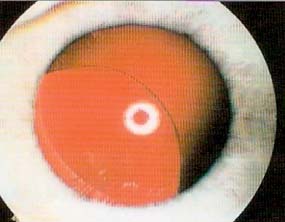
Figura 2 - Luxação inferior do cristalino
Na primeira consulta é de salientar a estatura superior ao P 95 com diminuição da razão entre os segmentos superior e inferior.
Qual o seu diagnóstico?
DISCUSSÃO
A homocistinúria clássica é causada pelo défice em cistationina β sintetase, enzima cujo cofactor é a piridoxina e que permite a conversão da homocisteína em cistationina. O gene CBS, que codifica esta doença autossómica recessiva, está localizado no braço longo do cromossoma 21. A frequência da homocistinúria clássica é variável, sendo a incidência estimada de 1/200.000 na nossa população.
É uma doença com início insidioso e envolvimento progressivo dos órgãos alvo nomeadamente olho (luxação do cristalino, miopia e glaucoma), esqueleto (membros longos, estatura alta e aracnodactilia que conferem aos doentes um fenótipo marfanoide), sistema vascular (tromboembolismo) e sistema nervoso central (atraso cognitivo e acidentes vasculares cerebrais). Ao nascimento a criança é normal, podendo a doença manifestar-se em qualquer idade desde a primeira infância á idade adulta. Embora em algumas crianças possa ser já evidente algum atraso de desenvolvimento psicomotor, o diagnóstico é efectuado geralmente após os dois anos de idade pelo envolvimento ocular, sendo com frequência o ofalmologista a alertar para o diagnóstico ao detectar a luxação do cristalino, que é evidente em 80% dos casos antes dos 10 anos de idade. A osteoporose está presente em praticamente todos os doentes, após a primeira infância levando a fracturas e escoliose. As alterações vasculares, caracterizadas por arteriosclerose prematura e complicações tromboembólicas das veias e artérias, são evidentes em 50% dos doentes antes dos 30 anos de idade condicionando o prognóstico desta doença pela sua elevada morbilidade e mortalidade. O atraso mental que é relativamente frequente (60 % dos casos), raramente é grave.
Os marcadores bioquímicos que orientam o diagnóstico são um aumento da homocistina e metionina com níveis reduzidos de cistina e cistationina no perfil de aminoácidos séricos e urinários. Este diagnóstico deverá ser confirmado pelo doseamento da enzima cistationina β sintetase em fibroblastos ou pelo estudo molecular. Estão descritas mais de 130 mutações nesta doença. Estas mutações determinam a resposta à piridoxina sendo a evolução mais favorável nos casos que respondem a esta vitamina. Na Península Ibérica predominam as mutações não respondedoras, principalmente a T191M frequente no norte de Portugal.
O objectivo do tratamento é reduzir os níveis de homocisteína total para níveis o mais próximo possível do normal. Cerca de 50% dos doentes respondem total ou parcialmente, à administração de doses suprafisiológicas de piridoxina. Nos casos em que não há resposta ou que a resposta é parcial o tratamento passa por uma dieta hipoproteica e pela administração de betaína (fármaco que cria uma via alternativa para eliminar a homocisteína). A suplementação com ácido fólico e vitamina B12 é também necessária.
Apenas um tratamento precoce iniciado nas primeiras semanas de vida permite uma evolução totalmente normal a longo prazo, justificando a inclusão desta doença no rastreio neonatal. Em Portugal, o rastreio sistemático da homocistinúria é feito desde 2005.
O aconselhamento genético deve ser efectuado a estas famílias e o diagnóstico pré natal é possível pelo estudo enzimático ou pelo estudo molecular em amniócitos ou nas vilosidades coriónicas.
BIBLIOGRAFIA
1. Couce ML, Fraga JM. Homocistinuria y alteraciones del metabolismo de folate e vitamina B12. In: P. Sanjurjo, A. Baldellou, editors. Diagnostico y tratamiento de las enfermedades metabólicas hereditarias. 4th ed. Madrid: Ergon 2006; 357-75. [ Links ]
2. Andria G, Fowler B, Sebastio G. Disorders of sulfur amino acid metabolism. In: Fernandes J, Saudubray JM, van den Berghe G, Walter JH editors. Inborn metabolic diseases: diagnosis and treatment. 5th ed. Berlin Heidelberg: Springer 2012; 311-22. [ Links ]
3. Linha Rara. Disponível em: http://www.linharara.pt/index.php?option=com_content&view=article&id=104&Itemid=26 [ Links ]

