










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.21 no.2 Porto jul. 2012
Incontinentia Pigmenti – caso clínico
Sónia Santos1, Rita S. Oliveira1, Vítor Bastos1, José Matos2, Isabel Andrade1
1 S. Pediatria, CH Tondela-Viseu
2 S. Dermatologia, CH Tondela-Viseu
RESUMO
Introdução: A Incontinentia pigmenti (IP) é uma rara genodermatose neuroectodérmica, com uma incidência de 1:50.000 nascimentos, sendo tipicamente letal no sexo masculino, in utero.
Caso Clínico: Os autores apresentam o caso de um recém-nascido, com lesões papulo-vesiculosas desde o nascimento sugestivas de IP. A evolução, a anatomo-patologia e o estudo genético permitiram estabelecer o diagnóstico.
Conclusão: A IP é uma entidade clínica potencialmente grave que requer um diagnóstico precoce e um seguimento multidisciplinar. A suspeita clínica é fundamental para chegar ao diagnóstico.
Palavras-chave: Incontinentia Pigmenti, recém-nascido, lesões papulo-vesiculosas.
Incontinentia pigmenti – a case report
ABSTRACT
Introduction: Incontinentia pigmenti (IP) is a rare genodermatosis neuroectodermal multisystem disorder. This disease has an incidence of 1:50.000 births and is typically lethal in males, in utero.
Case report: The authors present a clinical case of IP in a newborn with papulovesiculous eruptions observed on the first day of life. The evolution, anatomopathology and genetic study findings established the diagnosis.
Conclusion: The IP is a potentially serious clinical entity, which requires an early diagnosis and multidisciplinary follow-up.
Keywords: Incontinentia pigmenti, newborn, papulovesiculous eruptions.
INTRODUÇÃO
A Incontinentia Pigmenti (ou Síndrome de Bloch-Sulzberger) é uma genodermatose de transmissão dominante ligada ao cromossoma X(1), com uma incidência de 1:50.000 nascimentos(2), sendo habitualmente letal no sexo masculino, in utero(1). Foi descrita pela primeira vez por Bloch em 1926 e, posteriormente, por Sulzberger em 1928(1). As alterações cutâneas estão geralmente presentes ao nascimento e evoluem ao longo de quatro estadios: vesicular, verrucoso, hiperpigmentar e hipopigmentar(1). Em 50-80% dos casos há envolvimento extracutâneo: dentário, ungueal, ocular, neurológico ou ósseo(3). O diagnóstico é clínico(1), sendo confirmado pela biópsia cutânea. O estudo molecular complementa o estudo(4,5). Por se tratar de uma doença rara, descrevemos um caso cujas manifestações cutâneas estavam presentes ao nascimento e cujo follow-up tem demonstrado a ausência de envolvimento extra-cutâneo, apesar de expressa a mutação mais comum na IP.
CASO CLÍNICO
Recém-nascido (RN) do sexo feminino, de pais consanguíneos (primos em terceiro grau), fruto de uma terceira gestação (aborto espontâneo na primeira gravidez às sete semanas) vigiada e sem intercorrências. Nasceu às 40 semanas de gestação, por parto eutócico, com Apgar de 9/10 e peso, estatura e perímetro cefálico nos percentis 25,50 e 25, respectivamente. Logo ao nascimento foi visualizado um exantema papulo-vesiculoso com base eritematosa, de padrão linear a nível dos antebraços com generalização posterior, mas poupando a face (Figura 1), sendo o restante exame clínico sem alterações. Perante a suspeita de exantema infeccioso realizou estudo analítico (hemograma e proteína C reactiva) do qual se destacou apenas a presença de discreta eosinofilia (4,4%). As lesões foram-se generalizando, sempre com disposição linear, algumas com aspecto verrucoso (Figura 2). Teve alta ao sétimo dia de vida sob aleitamento materno, com rastreio auditivo normal e consultas de seguimento programadas (Neonatologia e Dermatologia). Os antecedentes familiares eram irrelevantes e o irmão, de nove anos, saudável.
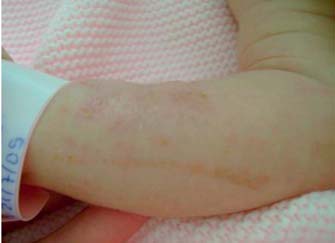
Figura 1 – Exantema papulovesiculoso de padrão linear, observado em D1 de vida.
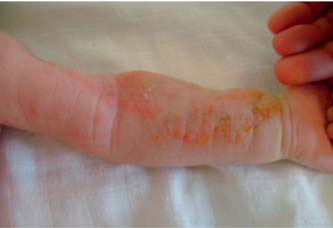
Figura 2 – Exantema nos antebraços com aspecto verrucoso.
Perante a evolução clínica foi colocada a hipótese de IP e ao 15º dia de vida foi realizada biópsia cutânea. O exame anatomopatológico revelou vesículas intra-epidérmicas e infiltrados de eosinófilos na derme papilar, confirmando o diagnóstico de Incontinentia Pigmenti em fase vesicular (Figura 3). A ecografia transfontanelar solicitada revelou alterações inespecíficas: pequenos focos hiperecogénicos nos gânglios da base, sugerindo microvasculopatia discreta. Foi pedida também a colaboração de Oftalmologia, cuja avaliação seriada foi normal.
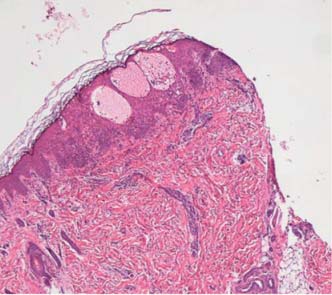
Figura 3 – Vesículas intra-epidérmicas e infiltrados de eosinófilos na derme papilar (gentilmente cedida pela Dra. Helena Garcia (CEDAP).
O estudo molecular revelou deleção nos exões quatro a dez no gene IKBKG/NEMO, sendo posteriormente pedido o estudo molecular à mãe que foi negativo. Aos três meses apresentava algumas lesões hiperpigmentadas nas coxas com padrão reticulado (Figura 4). Tem tido um desenvolvimento normal, sem manifestações extra-cutâneas até ao momento, nomeadamente oculares ou neurológicas.
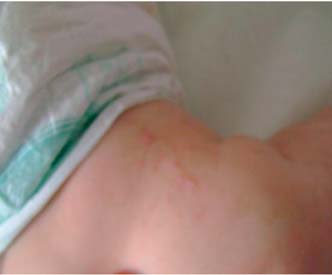
Figura 4 – Lesões hiperpigmentadas na coxa.
DISCUSSÃO
As manifestações cutâneas da IP estão geralmente presentes ao nascimento(1), como se verificou no caso descrito. A evolução destas, verifica-se ao longo de quatro estadios, cujo início e duração apresentam uma grande variabilidade individual(5). Todavia, a pessoa afectada pode não experimentar todos os estádios(5) ou estes podem até coexistir simultaneamente(6). O lactente apresentou as lesões cutâneas típicas dos três primeiros. O estadio I ou vesicular, apresenta-se sob a forma de vesículas lineares, pústulas e bolhas com eritema ao longo das linhas de Blaschko, presentes ao nascimento, mas que podem recorrer na infância ou nas intercorrências infecciosas febris(1). Acompanha-se frequentemente de eosinofilia(1), que pode atingir os 65%(7), embora no caso descrito a eosinofilia fosse ligeira. O estadio II ou verrucoso consiste em lesões verrucosas, com pápulas e placas queratóticas que ocorrem entre a 2ª e a 8ª semana de vida(1), desaparecendo por volta dos seis meses em mais de 80% dos casos(6). O estadio III ou de hiperpigmentação caracterizado por lesões maculares hiperpigmentadas ao longo das linhas de Blaschko, sobretudo nas virilhas, axilas e mamilos, ocorre entre a 12ª e a 40ª semana de vida(1) e desaparece por volta da segunda década de vida(7). Finalmente o último estadio de hipopigmentação, caracterizado por lesões lineares hipopigmentadas associadas ou não a lesões cutâneas atróficas, decorre desde a infância até à idade adulta(1).
Em 65-90% ocorrem anomalias dentárias e maxilares como atraso da erupção dentária, cáries, anodontia parcial, hipodontia, microdontia, micrognatia, prognatia, dentes cónicos(1). Em 40-60% pode surgir onicodistrofia ou displasia ungueal (e mais raramente tumores queratóticos sub-ungueais) (1) e em 35-70% alopécia do vertex(1). Em 30-60% podem surgir anomalias oculares (geralmente unilateral)(7): alterações pigmentares da retina, microftalmia, cataratas, leucocoria, estrabismo, atrofia óptica ou hipoplasia da fovea, glaucoma congénito, anomalias dos vasos periféricos da retina, cegueira(1). As anomalias do sistema nervoso central ocorrem em 10-40% dos casos(1) sendo variados: microcefalia (4%), atraso mental (12%), espasticidade (11%), convulsões (13%), ataxia, encefalopatia, hiperactividade, derrames cerebrais(7,8,9). Em 14% surgem anomalias ósseas (geralmente associadas a anomalias cerebrais graves) (1): hemivertebras, escoliose, espinha bífida, sindactilia, costelas supra-numerárias, deformidades do crânio. Em 1% podem surgir anomalias da mama: hipoplasia mamária ou mamilo supra-numerário(1) e até aplasia(7). Apenas o follow-up seriado desta criança poderá concluir pelo aparecimento das manifestações referidas.
O diagnóstico é feito com base na evolução das lesões cutâneas(1) e a confirmação faz-se por biópsia cutânea, correspondendo a cada um dos estadios alterações histopatológicas específicas(1). No caso descrito, revelou alterações típicas da fase vesicular. Cada estadio pode mimetizar outras doenças, impondo o respectivo diagnóstico diferencial, que no vesicular faz-se com: Impétigo bolhoso, Dermatose bolhosa, Pênfigo bolhoso, Eritema Tóxico, Dermatite Herpetiforme, Herpes Zoster(10) e Sífilis congénita(1,3); e no estadio IV faz-se com a Hipomelanose de Ito e com Vitiligo.
A neuroimagiologia, com recurso a Tomografia Axial Computorizada ou Ressonância Magnética Crânio-Encefálica, só se justifica quando ocorrem alterações no exame neurológico ou no desenvolvimento, convulsões ou vasculopatia da retina(1), o que até à data não ocorreu.
O estudo molecular revela maioritariamente, uma mutação no gene IKBKG/NEMO do cromossoma X, sendo encontrada deleção dos exões 4-10 em 60-80% dos casos(5,11,12), como se verificou neste. Quando não existe história familiar de IP, importa testar a mãe dada a grande variabilidade fenotípica, e caso a mutação esteja presente o aconselhamento genético é importante, pela possibilidade de diagnóstico pré-natal(5). Em mais de 65% dos casos estão reportadas mutações esporádicas(12). O gene referido intervém na protecção celular contra a apoptose celular(4), registando-se, desta forma, uma maior susceptibilidade a neoplasias (leucemia mielóide aguda, tumor de Wilms, retinoblastoma) (1).
Não existe tratamento específico(1). Na fase vesicular as lesões devem permanecer intactas e limpas para evitar a sobreinfecção bacteriana(1).
O follow-up destas crianças deve envolver uma equipa multidisciplinar: Oftalmologia, Neonatologia/Pediatria, Medicina Dentária, Genética e Neurologia (se alterações neurológicas)(1).
O prognóstico neste caso é bom, uma vez que não surgiram até à data, lesões oculares ou do Sistema Nervoso Central.
CONCLUSÃO
A IP, embora rara, exige um elevado índice de suspeição, pois devido aos seus diferentes estadios pode mimetizar diferentes doenças. Devido ao seu envolvimento extra-cutâneo, o follow-up por uma equipa multidisplinar torna-se crucial.
BIBLIOGRAFIA
1. Chang CH. Incontinentia Pigmenti. In: Emedicine 2007 Apr 6. Available at http://www.emedicine.com. Accessed Aug 2009. [ Links ]
2. Ehrenreich M, Tarlow MM, Godlewska-Janusz E, Schwartz RA. Incontinentia pigmenti (Bloch Sulzberger Syndrome): a systemic disorder. Cutis 2007; 79: 355-62. [ Links ]
3. Sordo ML, Vega BV, Zamora EH, Rosas AG, Pérez LA. Incontinentia pigmenti: informe de un caso familiar en varones afectados. Revisión de la literatura. Med Oral Patol Oral Cir Bucal 2005; 10: 122-29. [ Links ]
4. Roca AP, Baquero-Artigao F, Garcia-Miguel MJ, Vázquez JG, Fernández JG, Cuevas MV et al. Incontinentia pigmenti. Manifestaciones iniciales y a largo plazo. An Pediatr (Barc.) 2008; 68: 9-12. [ Links ]
5. Scheuerle A, Ursini MV. Updated October 28, 2010. Incontinentia Pigmenti (Bloch-Sulzberger syndrome). In: GeneReviews at GeneTests: Medical Genetics Information Resource. Copyright, University of Washington, Seattle. 1997-2011. Available at http://www.genetests.org. Accessed Decemb 2011. [ Links ]
6. Taborda A, Bastos V, Fonseca J, Ruas E, Moreno A. Incontinência Pigmenti – Caso clínico. Acta Pediatr Port 1999; 30: 499-502. [ Links ]
7. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet 1993; 30:53-9. [ Links ]
8. Castroviejo IP, Pascual SI, Fragua RV, Martinez V. Incontinentia pigmenti. Halllazgos clínicos y radiológicos en una serie de 12 pacientes. Neurologia 2006; 21(5): 239-48. [ Links ]
9. Maingay-de Groof F, Lequin MH, Roofthooft DW, Oranje AP, de Coo IF, Bok LA et al. Extensive cerebral infarction in the newborn due to incontinentia pigmenti. Eur J Paediatr Neurol 2008; 12: 284-9. [ Links ]
10. Faloyin M, Levitt J, Bercowitz E, Carrasco D, Tan J. All that is vesicular is not herpes: Incontinentia pigmenti masquerading as herpes simplex virus in a newborn. Pediatr 2004; 114: 270-2. [ Links ]
11. Fusco F, Fimiani G, Tadini G, D Urso M, Ursini MV. Clinical diagnosis of incontinentia pigmenti in a cohort of male patients. J Am Acad Dermatol 2007; 56:264-7. [ Links ]
12. Fusco F, Pescatore A, Bal E, Ghoul A, Paciolla M, Lioi MB et al. Alterations of the IKBKG locus and diseases: an update and a report of 13 novel mutations. Hum Mutat 2008; 29:595-604. [ Links ]
Sónia Santos
E-mail: soniafmuc@gmail.com

