










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.21 no.4 Porto dez. 2012
Osteogenesis imperfecta com manifestação pré-natal
Vinhas da Silva1, Ana Luísa Leite1, Jorge Sales Marques2, Márcia Gonçalves3, Manuela Mateus3
1 S. Pediatria, CH Vila Nova de Gaia Espinho
2 U. Genética, CH Vila Nova de Gaia Espinho
3 U. Neonatologia, CH Vila Nova de Gaia Espinho
RESUMO
Introdução: A osteogenesis imperfecta (OI) é uma doença genética rara, devida a alterações estruturais ou quantitativas do colagénio tipo 1. A classificação em tipos tem sofrido alterações recentes. O espectro clínico é variado, desde formas ligeiras (OI tipo I) ou moderadas (OI tipo IV-VII), até formas muito graves (OI tipo II-III).
Caso Clínico: É apresentado o caso de um recém-nascido do sexo masculino, fruto de uma primeira gestação, mal vigiada, com o diagnóstico de OI, com manifestações pré-natais (encurtamento dos membros inferiores e desmineralização da calote craniana na ecografia das 34 semanas). No exame objetivo, apresentava atividade espontânea diminuída, crânio grande com fontanelas alargadas e comunicantes, calote craniana fina e depressível, facies peculiar com nariz em sela, palato em ogiva, escleróticas brancas, membros curtos e com desvio anómalo. A radiografia do esqueleto ao nascimento apresentava múltiplas fraturas dos ossos longos dos membros. Apesar do início do tratamento com pamidronato, verificou-se agravamento clínico com surgimento de novas fraturas e insuficiência respiratória de agravamento progressivo e óbito.
Discussão: O caso clínico descrito parece corresponder a uma situação grave de OI tipo III ou tipo II, podendo ter hereditariedade autossómica dominante por mutação de novo ou autossómica recessiva, de acordo com a classificação atual, com manifestações muito precoces, pré e perinatais, com um desfecho fatal.
Palavras-chave: Osteogenesis imperfecta, pré-natal.
Prenatal presentation of osteogenesis imperfecta
ABSTRACT
Introduction: Osteogenesis imperfecta(OI) is a rare genetic disease, caused by generalized defect in collagen type 1. Clinical forms of the disease vary from mild (OI type I) or moderate (OI type IV-VII) to severe features (OI type II and III).
Case Report: We report a case of a male patient, born after an inadequate pregnancy surveillance, with prenatal presentation of OI (ultrasound performed at 34th gestational week revealed shortening of lower limbs and bone demineralization). Physical examination findings were frog-leg posture, macrocephaly, large anterior fontanel, white sclera, short limbs, tenderness and crepitation of limbs. Newborns X-rays showed numerous fractures. Although treatment with pamidronate was started, new fractures were detected. General conditions progressively aggravated and he died at 45th day of life.
Discussion: The reported case seems to be a severe form of type III or type II OI, with autosomal dominant transmission due to a de novo mutation or autosomal recessive transmission, according to current classification, with prenatal presentation and a fatal outcome.
Keywords: Osteogenesis imperfecta, prenatal.
INTRODUÇÃO
A osteogenesis imperfecta (OI) é uma doença genética rara, com uma incidência estimada de 1:10.000-20.000 nascimentos, devida a alterações estruturais ou quantitativas do colagénio tipo 1(1-4). Clinicamente é caracterizada por fragilidade esquelética e suscetibilidade a fraturas ósseas, espontâneas ou secundárias a traumatismos minor, baixa estatura, laxidez articular, alterações da dentição e perda da capacidade auditiva(1,5). O espectro clínico é variado, desde formas ligeiras (OI tipo I) ou moderadas (OI tipo IV-VII), até formas muito severas (OI tipo II-III)(1,5-8). A maioria dos casos é de transmissão autossómica dominante, existindo contudo formas autossómicas recessivas, de acordo com as novas classificações(8,9). O diagnóstico e classificação são baseados nas manifestações clínicas e radiológicas e na positividade da história familiar(1). A abordagem da doença deve ser multidisciplinar, envolvendo pediatras, ortopedista e fisiatra(1,2,4) sendo que a terapêutica é apenas de suporte. A utilização dos bifosfonados reduz a reabsorção óssea mediada pelos osteoclastos, parecendo reduzir a dor óssea e a incidência de fraturas(1,2,4).
CASO CLÍNICO
Recém-nascido do sexo masculino, fruto de uma primeira gestação de casal jovem, saudável e não consanguíneo, com vigilância irregular e tardia. Serologias maternas adequadas (não imune à toxoplamose, imune à rubéola, marcadores víricos negativos). Primeira ecografia pré-natal realizada às 34 semanas de idade gestacional com encurtamento dos membros inferiores e desmineralização da calote craniana. Realizou nesta altura estudo molecular para identificação de displasias ósseas (FGFR3, COL2A1, SLC26A2, CRTAP, LEPRE1, SOX9) que foi negativo, e ecocardiograma fetal sem alterações. O parto foi distócico por cesariana eletiva às 38 semanas. Índice de Apgar 6/8/9 com necessidade de reanimação com máscara e insuflador. Peso ao nascimento: 3140g (P50-75). O comprimento não foi avaliado devido às dificuldades na manipulação.
Sem história de doenças heredo-familiares conhecidas, nomeadamente displasias ósseas.
Admitido na Unidade de Cuidados Intensivos Neonatais por dificuldade respiratória. À admissão, hemodinamicamente estável (TA 65/45 mmHg, FC 130 bpm), em ventilação espontânea sem necessidade de oxigénio suplementar (SatO2 94%), bem perfundido, com choro vigoroso mas atividade espontânea globalmente diminuída; crânio grande com fontanelas alargadas e comunicantes, calote craniana fina e depressível; facies peculiar com nariz em sela, palato em ogiva e escleróticas brancas; tórax estreito e retração intercostal, auscultação cardíaca e pulmonar sem alterações; membros curtos e com desvio anómalo.
As radiografias do esqueleto revelaram a presença de múltiplas fraturas dos ossos longos dos membros (Figura 1), em diferentes estádios de evolução, com aspeto típico de popcorn. O estudo do metabolismo fosfo-cálcico foi normal.
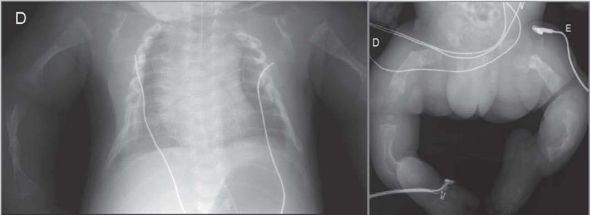
Figura 1 – Radiografia do esqueleto no primeiro dia de vida, com múltiplas fraturas dos ossos longos dos membros.
Foi instituído tratamento de suporte, com imobilização e estabilização das fraturas, e analgesia com paracetamol e morfina. Iniciou terapêutica com pamidronato na dose 0.5mg/kg ao trigésimo dia de vida, sem melhoria. Verificado agravamento progressivo com surgimento de novas fraturas espontâneas/mobilização minor (Figura 2), desmineralização óssea cada vez mais acentuada e insuficiência respiratória de agravamento progressivo, com óbito ao quadragésimo quinto dia de vida.
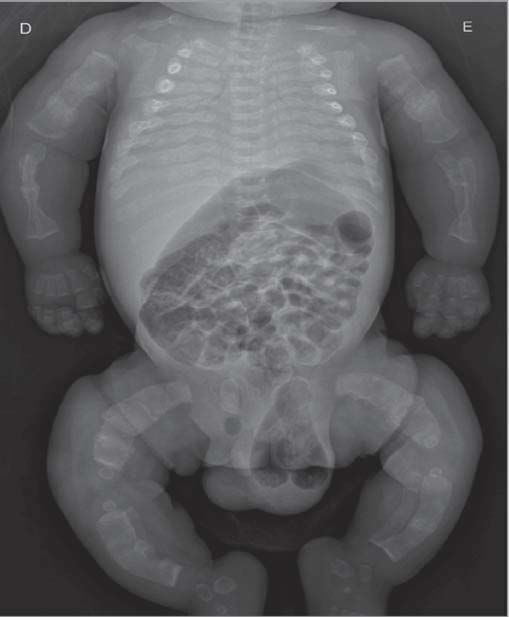
Figura 2 – Radiografia do esqueleto no quadragésimo dia de vida, com aparecimento de novas fraturas.
DISCUSSÃO
A OI apresenta um espectro clínico muito amplo, podendo ser classificada em quatro formas principais com características particulares que as distinguem, mas também com sobreposição das formas de apresentação e hereditariedade(1,5-8). O caso clínico descrito apresenta características clínicas e radiológicas das duas formas mais graves de OI (tipos II e III). A favor da OI tipo II apresenta múltiplas fraturas in utero com consolidação evidente no período pós-natal; extremidades curtas e com deformidade angular; fontanelas amplas. A favor da OI tipo III apresenta múltiplas fraturas presentes ao nascimento e deformidade óssea progressiva; macrocefalia com fácies triangular, escleróticas brancas e a sobrevida para além da primeira semana de vida(1,5).
O diagnóstico pré-natal, nomeadamente a ecografia obstétrica, é um elemento fundamental na avaliação da OI, sobretudo se realizada em centros de referência, uma vez que pode apontar para o diagnóstico antes das 20 semanas de gestação nas formas mais graves(2,4,10). Neste caso, a vigilância irregular da gestação conduziu a um diagnóstico tardio às 34 semanas, impossibilitando o recurso à interrupção médica, a ponderar pelo mau prognóstico da situação em causa.
O aconselhamento genético ao casal poderá ajudar na tomada de decisões pelo casal relativamente a futuras gestações, sendo neste caso dificultado pela incerteza em relação à forma de hereditariedade. O uso de técnicas para análise de DNA tem evidenciado a existência de doentes que, mesmo diagnosticados com a OI, não apresentam mutações nos genes responsáveis pela produção do colágeno.
A OI caracteriza-se por uma atividade osteoclástica aumentada e, consequentemente, um aumento da reabsorção óssea. A terapêutica com bifosfonados inibe a atividade osteoclástica e, de acordo com a literatura, quando utilizados na doença severa apresentam efeitos benéficos, nomeadamente na densidade óssea e diminuição da dor óssea(1,2,4). No caso em concreto não foi possível avaliar a sua utilidade dado o óbito pouco tempo após a sua introdução. Não parece, no entanto, ter ocorrido algum efeito sobre a dor, com todas as limitações decorrentes do pouco tempo de administração. A associação entre o primeiro ciclo de pamidronato e o aparecimento de sinais de dificuldade respiratória descrita por alguns autores(11), não parece adequar-se a este caso atendendo não existir patologia pulmonar prévia, bem como a ausência de resposta à terapêutica broncodilatadora.
BIBLIOGRAFIA
1. Rodriguez-Herrera G, Navrro-Charpantier MJ. Osteogénesis imperfecta con manifestaciones en el periodo neonatal. Acta méd. costarric 2009; 51:114-8. [ Links ]
2. John BM, Patnaik SK, Thergaonkar RW. Multiple fractures in neonates and osteogenesis imperfecta. MJAFI 2006; 62:73-4. [ Links ]
3. Paterson Colin R, McAllison Susan J. Classical Osteogenesis Imperfecta and Allegations of Nonaccidental Injury. Clin Orthop and Relat Reser 2006; 452:260-4. [ Links ]
4. Bastos F, Perez LT, Narváes C, Costa O, Silva R, Van-Dunem J, et al. Severe osteogenesis imperfecta: case report. Einstein 2010; 8:480-2. [ Links ]
5. Hoffman, Jodi D, Estrella E. Presentation of connective tissue disorders. NeoReviews 2007; 8:110-9. [ Links ]
6. Niyibizi C, Smith P, Zhibao M, Robbins P, Evans C. Potencial of gene terapy for treating osteogenesis imperfecta. Clin Orthop and Relat Reser 2009; 379:126-33. [ Links ]
7. Silence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Gent 1979; 16:101-6. [ Links ]
8. Van Dijk FS, Pals G, Van Rijn RR, Nikkels PG, Cobben JM. Classification of Osteogenesis imperfecta revisited. Eur J Med Genet 2010; 53:1-5. [ Links ]
9. Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet 2010; 155A: 943-68. [ Links ]
10. Morgan JÁ, Marcus PS. Prenatal diagnosis and management of intrauterine fracture. Obstet Gynecol Surv 2010; 65:249-59. [ Links ]
11. Munns CF, Rauch F, Mier RJ, Glorieux FH. Respiratory distress with pamidronate treatment in infants with severe osteogenesis imperfecta. Bone 2004; 35:231-4. [ Links ]
António Vinhas da Silva
E-mail: vinhasdasilva@gmail.com

