










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.22 no.1 Porto mar. 2013
Caracterização das infeções em doentes com Síndrome de deleção 22q11.2
Characterization of infections in patients with 22q11.2 deletion Syndrome
Manuel Oliveira1, Carla Teixeira1, Júlia Vasconcelos2, Esmeralda Neves2, Sílvia Álvares3, Margarida Guedes1, Laura Marques1
1 U. Infeciologia Pediátrica e Imunodeficiências, S. Pediatria, CH Porto
2 S. Imunologia, CH Porto
3 S. Cardiologia Pediátrica, CH Porto
RESUMO
Introdução: O Síndrome de deleção 22q11.2 (SD22q11.2) tem uma incidência de 1/2000 a 1/7000 nados-vivos. Caracteriza-se por um grau variável de imunodeficiência que predispõe a infeções, nomeadamente sinopulmonares.
Material e métodos: Estudo retrospetivo de 12 doentes, todos apresentando a del22q11.2 de novo, incidindo na caracterização imunológica e no tipo e número de infeções documentadas.
Resultados: No que respeita aos estudos imunológicos, um doente apresentava linfopenia T grave e linfopenia B com hipogamaglobulinemia associadas a Síndrome de Evans; dois doentes linfopenia T ligeira transitória; seis linfopenia T ligeira/moderada persistente e três estudo imunológico normal. A incidência média de infeções foi 0.5/ano/doente (1.1/ano/doente abaixo dos três anos de idade). As mais documentadas foram otite média aguda, pneumonia e bronquiolite.
Discussão: Encontrou-se um número baixo de infeções/ano/doente e estas ocorreram maioritariamente abaixo dos três anos de idade. As infeções sino-pulmonares foram as mais documentadas e a evolução geralmente benigna. O caráter transitório idade-dependente das alterações imunológicas e a normal função dos linfócitos, mais do que o grau de linfopenia T, parecem contribuir para este facto.
Palavras-chave: Imunodeficiência, infeções, Síndrome de deleção 22q11.2.
ABSTRACT
Background: The 22q11.2 deletion syndrome (SD22q11.2) has an incidence of 1/2000 to 1/7000 live births. It is characterized by a variable degree of immunodeficiency that predisposes to infections, especially sino-pulmonary.
Material and Methods: A retrospective study of 12 patients with del22q11.2 de novo was performed, focusing on the immunological characteristics and the type and number of documented infections.
Results: The immunological studies showed one patient had severe T lymphopenia T and B lymphopenia with hypogammaglobulinemia associated with Evans syndrome, two patients had transient mild T lymphopenia, six had mild to moderate persistent T lymphopenia and three presented a normal immunological study. The mean incidence of infections was 0,5/year/patient (1,1/year/patient under age three). The most frequent were acute otitis media, pneumonia and bronchiolitis.
Discussion: There was a low number of infections/year/patient, and these occurred mostly under the age of three years. The sino-pulmonary infections were the most documented and the evolution was generally benign. The transient and age-dependent nature of the immunological changes and the normal immune cell function, rather than the degree of T lymphopenia appear to contribute to this fact.
Keywords: immunodeficiency, infections, 22q11.2 deletion syndrome.
INTRODUÇÃO
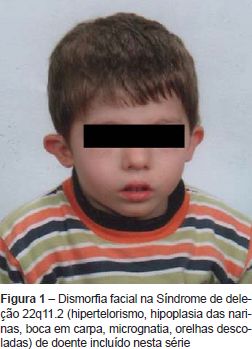
A Síndrome de deleção 22q11.2 (SD 22q11.2) é a designação genérica que engloba os doentes com Síndrome de DiGeorge (SDG), Síndrome velocardiofacial (SVCF) e Síndrome face-anomalia conotruncal (SFAC), substituindo a designação anterior de Síndrome CATCH 22 (Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate, Hypocalcemia)(1). A incidência é variável entre 1/2000 e 1/7000 nados-vivos. Tem por base uma deleção no braço longo do cromossoma 22 que origina haploinsuficiência dos genes afetados, identificada pela técnica de Fluorescence In Situ Hybridization (FISH) e ocorre em 90% dos doentes com SDG e 70-90% dos doentes com SVCF e SFAC(2,3). A transmissão é autossómica dominante em 20% dos doentes e 80% dos casos ocorrem de novo(2). A expressão fenotípica é variável, mesmo no contexto intrafamiliar; não existindo achados patognomónicos(4-6). Do espectro clínico fazem parte a dismorfia facial (Figura 1), as anomalias velo-faríngeas como fenda do palato aberta ou submucosa ou insuficiência velo-faríngea, as anomalias dos grandes vasos e malformações cardíacas [as mais específicas são as malformações conotruncais como a interrupção do arco aórtico tipo B, Truncus arteriosus, Tetralogia de Fallot, atrésia pulmonar e comunicação interventricular (CIV)]. A hipoplasia do timo é responsável por um grau variável de linfopenia T- SDG parcial e em <1% dos casos pode surgir Síndrome de imunodeficiência combinada severa por aplasia tímica- SDG completo. A hipoplasia das paratiroides origina hipocalcémia, sobretudo neonatal. Outras manifestações associadas a esta síndrome são dificuldades alimentares significativas, anomalias gastro-intestinais e genito-urinárias, atraso do crescimento e do desenvolvimento psicomotor, alterações do comportamento/doenças psiquiátricas, manifestações autoimunes. As alterações imunológicas, anatomo-funcionais velofaríngeas e défices nutricionais condicionam um maior risco infecioso nesta população de doentes.
OBJETIVOS
Quantificar e caracterizar as infeções dos doentes com SD22q11.2 seguidos atualmente em consulta de Pediatria do Centro Hospitalar do Porto.
MATERIAL E MÉTODOS
Estudo retrospetivo com base em dados demográficos, clínicos (incluindo número, tipo e evolução de quadros infeciosos bem documentados) e laboratoriais (estudo imunológico) recolhidos dos processos clínicos dos doentes. Incluíram-se aqueles com SD 22q11.2 e a deleção confirmada por FISH.
RESULTADOS
Foram incluídos 12 doentes, sete do sexo masculino (58%) e cinco do sexo feminino (42%). A idade média no diagnóstico foi de 41 meses (mínimo um mês, máximo 108 meses), apresentando um tempo de seguimento em consulta variável entre 1.5 e 16 anos (média de 8.3 anos). Um doente tinha antecedentes de um irmão falecido aos seis meses por cardiopatia congénita complexa e síndrome dismórfico de etiologia não esclarecida. Nos restantes, os antecedentes familiares eram irrelevantes. Em todos, o estudo molecular do doente e dos pais confirmou a deleção 22q11.2 de novo.
Seis doentes (50%) apresentavam cardiopatia congénita (dois casos com Tetralogia de Fallot e quatro com CIV, associada a atrésia pulmonar num deles, a persistência do canal arterial noutro e a arco aórtico direito com artéria subclávia esquerda aberrante noutro); cinco foram submetidos a cirurgia corretiva no primeiro ano de vida com evolução favorável; um apresenta quadro de hipertensão pulmonar arterial com necessidade de terapêutica farmacológica vasodilatadora pulmonar específica, no contexto de atrésia pulmonar com comunicação interventricular, artérias pulmonares não confluentes e hipoplásicas e colaterais aorto-pulmonares, sem solução operatória. Em nenhum doente se observou fenda do palato, apresentando um doente úvula bífida. Insuficiência velofaríngea foi descrita em seis doentes (50%), manifestada por dificuldades na articulação da fala e rinolália e/ou dificuldades alimentares com engasgamentos frequentes. Estes doentes foram orientados em terapia da fala e consultas de especialidade e uma doente foi submetida a faringoplastia de Orticochea aos sete anos de idade por incompetência velofaríngea marcada. Cinco doentes foram submetidos a cirurgia do foro ORL por infeções das vias aéreas superiores (adenoamigdalectomia e/ou miringotomia) e um doente a cirurgia plástica para correção de orelhas descoladas. A hipocalcémia por hipoparatiroidismo foi documentada num doente, sintomática nos primeiros três anos de vida por episódios de tetania com necessidade de terapêutica. O crescimento foi deficitário (peso e/ou comprimento/estatura inferior ao percentil 5) em seis doentes (50%) (quatro nos primeiros dois a cinco anos de vida), tendo sido os doentes avaliados em consulta de Gastrenterologia e Nutrição por má evolução ponderal e dificuldades alimentares. Nove doentes apresentavam atraso do desenvolvimento psicomotor, pontuando abaixo da média em avaliações psicométricas dos quais quatro com evidência de maior atingimento da área da linguagem.
Em relação à caracterização imunológica encontramos os seguintes resultados (Tabela 1): um doente (8%) apresentava linfopenia T grave persistente, associadamente foi-lhe diagnosticada uma púrpura imune e posteriormente Síndrome de Evans (caso 1); seis doentes (50%) apresentavam linfopenia T ligeira a moderada persistente (casos 2 a 7); dois (17%) linfopenia T ligeira transitória, um até aos 12 meses e outro até aos 18 meses de idade (casos 8 e 9); os restantes três doentes (25%) não apresentavam alterações no estudo imunológico (casos 10 a 12). Um doente realizou estudo funcional dos linfócitos e neste a expressão de marcadores de ativação dos linfócitos ex vivo e após estimulação in vitro, assim como o índice de blastogénese, estavam dentro de valores normais. À exceção do primeiro doente referido, nenhum outro apresentou manifestações autoimunes. Apenas este doente foi submetido a profilaxia com Cotrimoxazol e terapêutica de substituição com gamaglobulina endovenosa.
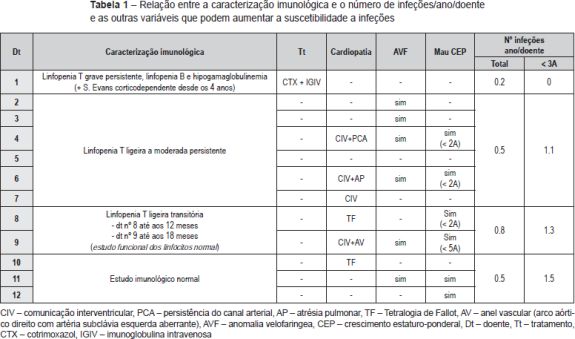
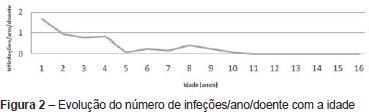
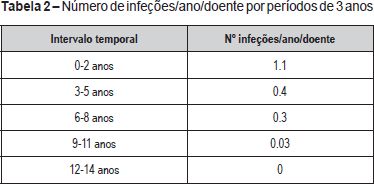
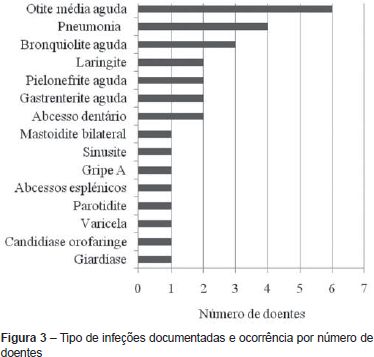
O número médio de infeções/ano/doente foi de 0,5, tendo-se verificado uma incidência maior abaixo dos 36 meses de idade (média de 1,1 infeção/ano/doente) e uma tendência decrescente com o passar dos anos (Figura 2 e Tabela 2). As infeções mais documentadas foram otite média aguda (OMA), pneumonia e bronquiolite (Figura 3), e a evolução foi geralmente favorável. Das intercorrências infeciosas documentadas na totalidade dos doentes, em dez episódios houve necessidade de internamento e/ou antibioticoterapia endovenosa. As duas intercorrências consideradas mais graves foram: um episódio de otomastoidite bilateral no contexto de OMA supurada que ocorreu aos 34 meses de idade numa doente com linfopenia T moderada, com evolução favorável; e o aparecimento de abcessos esplénicos de etiologia desconhecida, em contexto de infeção documentada a H1N1, e que resolveram com tratamento conservador, no doente com défice imunitário mais grave. Não se verificou nenhuma tendência aparente que sugerisse relação entre o grau de linfopenia e a incidência de infeções, nem com as restantes variáveis que podem influenciar a ocorrência destas (cardiopatia, anomalia velo-faríngea, atraso de crescimento) (Tabela 1).
DISCUSSÃO
O número de infeções/ano/doente encontrado foi baixo e estas ocorreram maioritariamente abaixo dos três anos de idade.
A otite média aguda, pneumonia e bronquiolite foram aquelas registadas com maior frequência e a evolução das intercorrências infeciosas foi geralmente benigna. O padrão encontrado é semelhante ao esperado numa população sem imunodeficiência. Em termos de literatura científica, os estudos que englobam doentes com o SD22q11.2 (excluindo a situação rara e particular dos casos de SDG completo) apontam para a ocorrência de infeções sino-pulmonares de repetição na primeira infância, nomeadamente sinusite, otite e broncopneumonia; embora facilmente tratáveis(4-6). A evolução clínica favorável justifica-se pelo facto de se encontrar associado à síndrome uma linfopenia T geralmente ligeira a moderada e transitória(7,8), verificando-se nos doentes com SDG parcial um aumento da população de células T memória ou efectoras com a idade(9). Por outro lado, a resposta proliferativa dos linfócitos a mitogénios e antigénios é normal, mostrando uma atividade funcional intrínseca não alterada(2,7,8). Assim, o estudo funcional dos linfócitos é mais indicativo da gravidade do defeito imunológico do que as contagens absolutas de células T(2). Na nossa amostra não se encontrou relação direta entre a gravidade da linfopenia e a ocorrência de infeções. Um dos doentes realizou estudo funcional dos linfócitos que se mostrou normal. Os doentes podem igualmente apresentar alterações inespecíficas da imunidade humoral (hipogamaglobulinemia; défice seletivo de IgA, défice de anticorpos específicos ou de subclasses de IgG)(7,8).
Para os peritos do International 22q11.2 Deletion Syndrome Consortium a caracterização imunológica (hemograma com diferencial e citometria de fluxo) deve ser efetuada na altura do diagnóstico e monitorizada de forma alargada (incluindo imunoglobulinas e função de células T) aos nove e 12 meses de idade (antes da administração de vacinas vivas)(10). Isto permite identificar doentes com maior risco infecioso e avaliar a necessidade de profilaxia antibiótica (geralmente não indicada) com base na contagem de linfócitos T(2). Na nossa série apenas um doente necessitou de profilaxia com cotrimoxazol por contagem de células T CD4+ <500/mm2 e de terapêutica de substituição com IgG endovenosa por linfopenia B e hipogamaglobulinemia pós-terapêutica com Rituximab no contexto de Síndrome de Evans. Em relação às imunizações, as vacinas vivas são seguras em doentes com idade superior a 12 meses e estudo funcional dos linfócitos normal, contagens de células T CD4+ >500/mm3 e CD8+>300/mm3 (7,8). Outros aspetos a ter em conta nos lactentes com SD 22q11.2 é a minimização da exposição a infeções, a vacinação anti-influenza, profilaxia para o vírus sincicial respiratório e a utilização de derivados de sangue irradiados (CMV-negativo) quando necessário(10).
As alterações anatomo-funcionais velofaríngeas, os défices nutricionais provocados pelas dificuldades na alimentação e a cardiopatia subjacente, além de serem a principal causa de morbilidade nestes doentes, podem também condicionar aumento da suscetibilidade a infeções(2). Na nossa amostra, o baixo número e pequena gravidade das infeções encontradas não nos permite tirar conclusões quanto à relação destas com as variáveis descritas. No entanto estas situações devem estar presentes na mente dos profissionais de saúde, permitindo estabelecer um plano adequado de cuidados antecipatórios para estes doentes.
BIBLIOGRAFIA
1. Almeida R, Alvares S, Fortuna A, Moreira J, Vieira A. Associação de Arco Aórtico Cervical a Delecção 22q11- Papel da RMN no Diagnóstico. Rev Port Cardiol 2003; 22:1241-8. [ Links ]
2. Mastroiacovo P, Rossi P, Cancrini C, Azzari C, DiGilio MC, Marino B, et al. Chromosome 22q.11 deletion - Recommendations for Diagnosis and Treatment. Italian Primary Immunodeficiencies Strategic Scientific Committee 2005. Disponível em: http://www.aieop.org/stdoc/prot/rec_del22_en_06.pdf [ Links ]
3. Koshiyama DB, Rosa RM, Zen PR, Pereira VL, Graziadio C, Coser VM, et al. Síndrome de deleção 22q11.2: importância da avaliação clínica e técnica de FISH. Rev Assoc Med Bras 2009;55:442-6. [ Links ]
4. Fernández L, Nevado J, Santos F, Heine-Suñer D, Martinez-Glez V, García-Miñaur S, et al. A deletion and a duplication in distal 22q11.2 deletion syndrom region. Clinical implications and review. BMC Med Genet 2009;10:48. [ Links ]
5. Bassett AS, Chow EWC, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet A 2005;138:307-13. [ Links ]
6. Hay BN. Deletion 22q11: spectrum of associated disorders. Semin Pediatr Neurol 2007;14:136. [ Links ]
7. Seroogy CM. DiGeorge syndrome: Evaluation, diagnosis, and management. Disponível em: http://www.uptodate.com/contents/digeorge-syndrome-evaluation-diagnosis-and-management [ Links ]
8. Seroogy CM. DiGeorge syndrome: Pathogenesis, epidemiology, and clinical manifestations. Disponível em: http://www.uptodate.com/contents/digeorge-syndrome-pathogenesis-epidemiology-and-clinical-manifestations [ Links ]
9. McLean-Tooke A, Barge D, Spickett GP, Gennery AR. Immunologic defects in 22q11.2 deletion syndrome. J Allergy Clin Immunol 2008;122:362-7. [ Links ]
10. Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, et al. Practical Guidelines for Managing Patients with 22q11.2 Deletion Syndrome. J Pediatr 2011;159:332-9. [ Links ]
Laura Marques

