










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.22 no.1 Porto mar. 2013
Parotidite recorrente juvenil nem sempre é o que parece
Juvenile recurrent parotiditis not always what it seems
Aida Silva e Sá1, José Carlos Fraga1, Ana Margarida Costa1, Fátima Dias2, Iva Brito3
1 S. Pediatria, CH Trás-os-Montes de Alto Douro
2 S. Reumatologia, CH São João
RESUMO
Introdução: A parotidite recorrente juvenil (PRJ) é uma inflamação recidivante idiopática da glândula parótida, geralmente associada a sialectasia não-obstrutiva. A síndrome de Sjögren (SS), rara em idade pediátrica, pode apresentar-se nesta faixa etária sob a forma de parotidites de repetição.
Caso clínico: Menina, de 13 anos, que no quarto episódio de tumefação e dor parotídea, iniciou investigação etiológica que revelou glândulas parótidas ecograficamente com textura heterogénea e múltiplas calcificações punctiformes, título ANA elevado (1:1280) com padrão mosqueado, anticorpos Anti-SSA e Anti-SSB positivos, assim como o fator reumatóide, híper-gamaglobulinémia e elevação da imunoglobulina G. Verificou-se, também, elevação da creatinofosfoquinase, da transamínase glutâmico-oxalacética e da transamínase glutâmico-pirúvica. A biópsia das glândulas salivares minor foi compatível com diagnóstico de SS. Iniciou terapêutica com hidroxicloroquina e corticóide oral em baixa dose com resposta clínica favorável.
Discussão/Conclusão: A idade de aparecimento da tumefação parotídea ajuda no diagnóstico diferencial entre PRJ idiopática e uma etiologia inflamatória crónica. O envolvimento muscular está descrito no SS primário e tem um espectro clínico e patológico variado, sendo que a miosite subclínica surge numa percentagem que varia entre os 5% e os 37%.
Palavras-chave: Hidroxicloroquina, miosite, parotidite recorrente, síndrome Sjögren.
ABSTRACT
Introduction: Juvenile recurrent parotiditis (JRP) is a relapsing inflammation of the parotid gland, usually associated with non-obstructive sialectasis. The Sjögren syndrome (SS), rare in children, may present at this age, in the form of recurrent parotiditis.
Case Study: Female, 13 years, in the fourth episode of parotid swelling and pain, begins investigating: ultrasound of the parotid glands revealed heterogeneous texture and multiple punctiforme calcifications, high ANA (1:1280) title, with speckled pattern, auto-antibodies anti-SSA, anti-SSB and Rheumatoid Factor positive, hyper-gamaglobulinemia and elevated Immunoglobulin G. We also observed elevation of Creatinofosfoquinase, Glutamic Oxaloacetic Transaminase and Glutamic Pyruvic Transaminase. A minor salivary gland biopsy was consistent with a diagnosis of SS. Treatment was started with systemic steroids combining hydroxychloroquine during exacerbations, with clinical improvement.
Discussion/conclusion: The age of onset of parotid swelling helps in differentiation between JRP idiopathic and other clinical entities. The muscle involvement occurs in primary SS and has a varied clinical and pathological spectrum, subclinical myositis occurs in a percentage that varies between 5% and 37%.
Keywords: Hydroxychloroquine, myositis, recurrent parotitis, Sjögren Syndrome.
INTRODUÇÃO
O aumento recorrente do volume das glândulas salivares é pouco comum na infância e representa um desafio diagnóstico e terapêutico na prática clínica(1).
São várias as hipóteses etiológicas que poderão ser colocadas perante um quadro de parotidite recorrente, como se mostra na Tabela 1.

A parotidite recorrente idiopática (PRI), juntamente com a parotidite epidémica, são as doenças inflamatórias das glândulas salivares mais comuns em idade pediátrica(2). A PRI consiste na inflamação recorrente da glândula parótida, geralmente associada a sialectasia não-obstrutiva, e manifesta-se por episódios recidivantes de aumento de volume e/ou dor na glândula parótida, uni ou bilateral, geralmente acompanhados de febre e mal-estar geral. É frequente a eliminação de secreção mucopurulenta pelo ducto de Stensen e diminuição do fluxo salivar(3). A idade de início é variável entre os oito meses e os 16 anos e resolve, na maioria dos casos, de forma espontânea após a puberdade(4). O número de crises inflamatórias agudas varia de doente para doente, embora o mais frequente seja a ocorrência de recidiva a cada três a quatro meses. A frequência dos surtos geralmente é mais elevada no primeiro ano escolar e tende a diminuir gradualmente até à puberdade(4). A ausência de novos episódios no período pós-pubertário poderá significar que os episódios recorrentes na infância se devam a imaturidade imunológica(5).
Embora mais raros, os quadros obstrutivos como cálculos, neoplasias ou malformações devem ser excluídos, assim como a existência de uma associação familiar que nos faça suspeitar de uma causa genética(6).
Alguns casos de parotidite recorrente estão associados a imunodeficiências, como o défice de imunoglobulina A (IgA) e infeção pelo vírus da imunodeficiência humana (VIH) (6).
Raramente, a parotidite pode ser a manifestação inicial da síndrome de Sjögren (SS). A SS é uma entidade rara em idade pediátrica com menos de 130 casos descritos na literatura científica(7), embora alguns autores afirmem que está, provavelmente, subdiagnosticada(7,8). Apenas 5% dos pacientes com diagnóstico com SS experimentam sintomas inaugurais antes dos 12 anos de idade(9). É uma doença crónica auto-imune, caracterizada por infiltrado linfocitário das glândulas exócrinas. Na criança pode apresentar-se sob a forma de parotidites de repetição e os sintomas de secura das mucosas são menos frequentes que na idade adulta(9). A etiologia do SS é complexa e inclui fatores genéticos de predisposição e, possivelmente, um trigger infecioso. Genes reguladores da apoptose influenciam a cronicidade do infiltrado linfocitário(10). Foram já propostos critérios para o diagnóstico de SS primário em idade pediátrica(11).
CASO CLÍNICO
Menina, com 13 anos de idade, de raça caucasiana, previamente saudável enviada à consulta externa de Pediatria do Centro Hospitalar de Trás-Montes-Alto-Douro por parotidites recorrentes. Dos antecedentes familiares a realçar patologia cardiovascular (pai falecido aos 37 anos por enfarte agudo do miocárdio e tio paterno com arritmia cardíaca não especificada).
Entre os 11 e os 13 anos de idade teve três episódios de tumefação e dor localizada no ângulo da mandíbula direita sem febre. Recorreu ao médico assistente, e em todos os episódios foi atribuído o diagnóstico clínico de parotidite sendo medicada com amoxicilina/ácido clavulânico e anti-inflamatório, com melhoria clínica. Em Agosto de 2009 (13 anos e quatro meses), recorreu ao Serviço de Urgência de Pediatria Médica do Centro Hospitalar de Trás-Montes-Alto-Douro com a mesma sintomatologia, agora à esquerda. A paciente negava febre, xerostomia, secura ocular, queixas articulares ou musculares, assim como sinais ou manifestações cutâneas. O exame objetivo, na admissão, não revelava alterações para além de tumefação parotídea esquerda que apagava o ângulo da mandíbula, de limites mal definidos e dolorosa no bordo superior. Foi, então, orientada para a consulta externa de Pediatria.
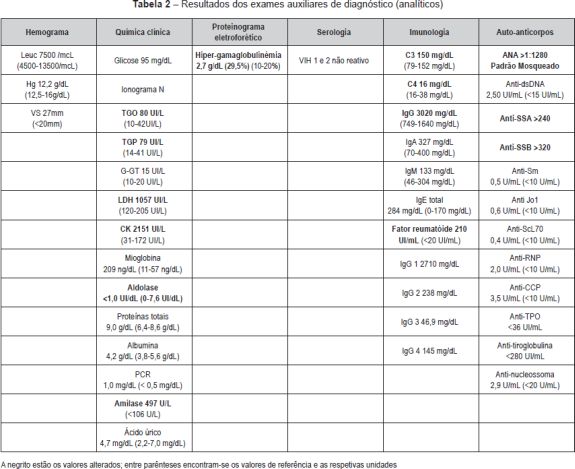
Os exames complementares realizados revelaram leucograma com 7500 leucócitos, hemograma com 12,2 g/dL de hemoglobina, velocidade de sedimentação de 27 mm (<20mm) e proteína C-reactiva de 1,0 mg/dL (< 0,5 mg/dL); valor de transamínase glutâmico-oxalacética (TGO) de 80 UI/L (10-42UI/L) e da transamínase glutâmico-pirúvica (TGP) de 79 UI/L (14-41UI/L) com gama-glutamil transpeptidase (G-GT) dentro dos valores da normalidade; lactato desidrogenase (LDH) de 1057 UI/L (120-205 UI/L) e creatininofosfoquinase (CK) de 2151 UI/L (31-172 UI/L), com valor de aldolase (<1) normal. Os valores de glicose, ionograma, proteínas totais, albumina e ácido úrico eram normais. O valor da amilase (497 U/L) estava elevado (N<106 U/L). O proteinograma eletroforético revelou hipergamaglobulinémia de 29,5% (2,7 g/dL). A ecografia das parótidas mostrou glândulas de contornos regulares e textura marcadamente heterogénea, com múltiplas calcificações punctiformes e alguns pequenos nódulos hipoecóides sugestivos de gânglios, bilateralmente, e várias formações ganglionares da região submandibular, as maiores pericentrimétricas. Na progressão da investigação etiológica as serologias víricas para o VIH 1 e 2 revelaram-se negativas. Os valores de C3 e C4 eram normais assim como os valores de IgM e IgA. O doseamento de IgG foi de 3020 mg/dL (749-1640 mg/dL). O fator reumatóide estava elevado (210 UI/mL). Rela tivamente ao estudo de auto-anticorpos a referir título de ANA elevado (1:1280), com padrão mosqueado e anti-SSA (>240) e anti-SSB (>320) também elevados; restantes auto-anticorpos (incluindo anti-dsDNA e anti-RNP) negativos (Tabela 2). O teste de Schirmer mostrou-se sem alterações ao final de cinco minutos. A biópsia de glândulas salivares minor do lábio inferior mostrou lesões de sialadenite crónica ao nível das glândulas salivares minor compatível com o diagnóstico clínico de SS. A eletromiografia não revelou alterações compatíveis com miosite/padrão miopático.
A ecografia cardíaca e o Holter revelaram-se dentro da normalidade. Foi medicada com ibuprofeno 600 mg/dia bid registando-se melhoria clínica ao final de oito dias. Iniciou hidroxicloroquina na dose de 310 mg numa toma única diária, em Outubro de 2009 que manteve como terapêutica de base. Manteve-se assintomática até Dezembro de 2009 altura em que recorreu novamente ao serviço de urgência do CHTMAD com tumefacção e dor parotídea à direita associada a gonartrite bilateral e omalgia. Associou corticóide sistémico, per os, (prednisolona 10 mg/dia) com melhoria progressiva dos sintomas. Em Março de 2010 recorreu a cuidados médicos com a mesma sintomatologia, tendo respondido favoravelmente ao mesmo esquema terapêutico com corticóide.
Já em seguimento em consulta externa de Reumatologia Pediátrica, por constatação nas análises de controlo de aumento das enzimas musculares (aldolase 16, LDH 300 IU/L, CK 600 IU/L), embora sem fraqueza muscular ou alterações cutâneas efetuou novo eletromiograma que não revelou alterações e biópsia de pele e músculos. A primeira revelou discreto infiltrado inflamatório crónico perivascular . A biópsia muscular não evidenciou fibras em necrose, infiltrados inflamatórios ou fibrose intersticial nem substituição adiposa, com estrutura interna das fibras normal.
Mantém-se assintomática desde Março de 2010, medicada diariamente com hidroxicloroquina.
DISCUSSÃO
Na investigação inicial de um doente com parotidites recorrentes, a ecografia e estudo analítico permitiram excluir as causas obstrutivas, infeciosas, genéticas bem como a presença de uma imunodeficiência. Perante isto, foram colocadas as seguintes hipóteses diagnósticas: SS com miosite, síndrome de overlap e dermatomiosite/polimiosiste.
A SS é uma doença auto-imune sistémica que, como referido, afeta predominantemente as glândulas exócrinas, sendo que a secura da mucosa ocular (queratoconjuntivite sicca) e a xerostomia constituem os sintomas clássicos da doença. Contudo, em idade pediátrica, a parotidite recorrente representa a manifestação oroglandular mais frequente(9). Outras manifestações não exócrinas incluem a vasculite- pele, músculo e articulações, serosas, sistema nervoso central (SNC) e periférico (SNP), doença vascular não inflamatória (fenómeno de Raynaud), doença imuno-mediada (citopenias, fadiga e febre), e endocrinopatia auto-imune (tiroidite) (10).Os achados laboratoriais e patológicos (infiltrado linfocitário das glândulas exócrinas, hipergamaglobulinémia, elevação da VS, título de anticorpos antinucleares (ANA) elevado com auto anticorpos anti-SSA e anti-SSB positivos) são comuns na idade pediátrica(8).
A idade de aparecimento da tumefação parotídea é, nos pacientes com SS, geralmente após os seis anos, já na PRI é antes esta idade(10). A SS pode surgir isolada (SS primária) ou associada a outras patologias reumatismais (SS secundária) sendo as mais frequentes o lúpus eritematoso sistémico, a esclerodermia e a doença mista do tecido conjuntivo(10).
Os critérios de diagnóstico de SS (adulto) propostos em 2005 pelo Grupo de Consenso Americano – Europeu não são adequados para a idade pediátrica. Os critérios de diagnóstico propostos para o SS primário em idade pediátrica estão descritos na (Tabela 3). O diagnóstico de SS baseia-se na presença de quatro ou mais dos 11 critérios apresentados(11).

Na paciente apresentada, a presença de aumento do volume das parótidas (critério 2), a presença de artralgias (critério 6), a demonstração de um título de ANA elevado (1:1280) com padrão mosqueado e Anti-SSA, Anti-SS-B e FR positivos (critério 7), a presença de alterações analíticas características com elevação da amílase sérica, da VS, a hipergamaglobulinémia e elevação da IgG (critério 8) associado a um padrão ecográfico das glândulas parotídeas e biópsia das glândulas salivares minor característicos (critério 10 e 11), permitiram o diagnóstico de SS.
Por outro lado, a negatividade dos auto-anticorpos para além dos anti-SSA e anti-SSB (nomeadamente o anti-RNP, anti-DsDNA, anti-Scl, anti-Jo1) tornam improvável, neste momento, a presença de outras patologias reumatismais associadas, como a doença mista do tecido conjuntivo, lúpus eritematoso sistémico ou esclerodermia.
Contudo, não podemos esquecer que várias doenças reumáticas podem compartilhar alterações serológicas(12,13). Isto é particularmente verdadeiro para anticorpos antinucleares, fator reumatóide, anticorpos anti-citrulina peptídeo (anti-CCP), e anti-Ro/SSA e anti-La/SSB(12,13). Nesta paciente foi detetada a positividade do anti-SSA e anti-SSB, mas não podemos esquecer que a positividade de ambos (mas principalmente o anti-SSA) pode ocorrer no LES. Assim, embora neste momento a paciente cumpra os critérios de diagnóstico de SS, esta é uma doença que em cerca de 50% dos casos precede por vários anos uma doença autoimune associada, e como tal, o diagnóstico da síndrome de overlap não pode ser completamente posto de parte.
O envolvimento muscular no SS primária tem um espectro clínico e patológico variado(14). Um terço dos doentes tem mialgias e/ou fraqueza muscular por vezes incapacitante(14,15). No estudo efetuado por Vrethem et al. foi reportada uma maior prevalência (73%) de achados inflamatórios na biópsia muscular de pacientes assintomáticos com SS primária. Uma miosite sintomática é descrita em 3% dos casos. A elevação de IgG parece estar associada a maior probabilidade de desenvolver miosite sintomática. A miosite subclínica é mais frequente como no presente caso e, consoante as diferentes séries, surge numa percentagem que varia entre os 5% e os 37%(14).
A associação entre SS e dermatomiosite/polimiosite poderia ser colocada tendo em conta a elevação das enzimas musculares. Segundo Bohan & Peter para a definição clínica de dermatomiosite/polimiosite têm de estar presentes associações de pelo menos três dos seguintes achados: manifestações cutâneas (eritema orbitário em heliotropo, pápulas de Gottron em superfícies extensoras), fraqueza muscular proximal e simétrica, elevação das enzimas musculares (CK, aldolase, DHL, TGO, TGP), alterações eletromiográficas típicas (unidades motoras polifásicas curtas, fibrilhações, ondas positivas agudas, irritabilidade de inserção e descargas bizarras ou repetitivas de alta frequência) e biópsia muscular com alterações típicas (evidência de necrose de fibras musculares, fagocitose, regeneração com basófilos, grandes vacúolos no sarcolema e nucléolo proeminente, atrofia de distribuição perifascicular, fibras musculares de tamanho variável e exsudado inflamatório perivascular), sendo que esta última é efetuada na presença de sintomas clínicos(7,10).
Nesta paciente temos, apenas, a elevação das TGO, TGP, CK, LDH e da aldolase, o que torna o diagnóstico de dermatomiosite/polimiosite improvável. Além disso, segundo alguns autores, a miosite como manifestação extraglandular da SS tende a ser caracterizada como menos severa e com uma resposta mais favorável à terapêutica com baixas/moderadas doses de corticoide comparado com a dermatomiosite/ polimiosite(14,15). Esta resposta favorável foi verificada nesta paciente.
Assim, por tudo o que foi descrito, parece-nos que o diagnóstico da paciente é SS com miosite.
Relativamente ao tratamento instituído na paciente é de referir que perante uma suspeita de PRI o tratamento deve ser conservador, com uso de analgésicos e orientações de cuidados locais (massagem, calor local e higiene bucal) sendo que, até ao presente, não se conhecem medidas preventivas para evitar a recorrência dos episódios. A prescrição de antibióticos é, geralmente, desnecessária se não houver suspeita de infeção secundária bacteriana (febre e/ou secreção purulenta). Nos casos de suspeita de parotidite bacteriana, deve-se instituir tratamento que permita cobertura do Streptococcus pneumoniae e Haemophilus influenzae(6).
Quanto ao tratamento do SS, os anti-inflamatórios não esteróides, corticosteróides e hidroxicloroquina são as drogas mais frequentemente utilizadas. No caso da paciente apresentada o tratamento com hidroxicloroquina associando agentes anti-inflamatórios nas agudizações permitiu mantê-la assintomática. O tratamento passa, também, pela terapêutica sintomática com lágrimas artificiais e fluidos para diminuir a secura das mucosas, medidas não instituídas na paciente uma vez que não tem queixas de secura das mucosas. Drogas imunossupressoras mais potentes, como a ciclosporina e ciclofosfamida, estão reservadas para o tratamento das manifestações mais graves(9,10,16,17).
Embora a SS primária tenha uma evolução benigna não podemos esquecer que a evolução desta patologia é crónica e insidiosa, e como já referido, poderá preceder uma doença auto-imune associada por vários anos. Devido à proliferação monoclonal de células B há um risco aumentado de linfoma de MALT, e também está descrito o aumento de risco de Linfoma não-Hodgkin pelo que a vigilância a longo prazo é fundamental(15,16).
BIBLIOGRAFIA
1. Piltcher O. Afecções das glândulas salivares na infância. In: Sih T, Chinski A, Eavey R, Godinho R. V Manual de otorrinolaringologia pediátrica da Interamerican Association of Pediatric Otorhinolaryngology (IAPO). 1.ª Edição. Brasil: Lis Gráfica e Editora;2006:p127-32. [ Links ]
2. Gajendra V, Nagabhushana D, Mamatha G, Annigeri R. Bilateral Parotid Swelling in a child – A case report. Indian Soc Pedodon Prev Dent 2004; 22:17-20. [ Links ]
3. Chitre V, Premchandra J. Recurrent parotitis. Arch Dis Child 1997;77:359-63. [ Links ]
4. Miziara ID, Campelo S. Parotidite recorrente da infância: estudo em longo prazo de cinco casos e revisão da literatura. Rev Bras Otorrinolaringol 2005;71:570-5. [ Links ]
5. Oliveira W, Assis L, Crosara S, Becker H, Guimarães R, Oliveira J et al. Síndrome de Sjögren primário em crianças: relato de caso. Rev Bras Cirurgia da Cabeça e Pescoço 2005;34:50-2. [ Links ]
6. Bricks LF. Parotidite recorrente da infância. Rev Paulista de Pediatria 2007;25: 190-2. [ Links ]
7. Woo P, Laxer M, Sherry D. Juvenile Dermatomyositis. Pediatric Rheumatology in Clinical Practice. 1st Edition. London: Springer Editors;2007:66-76. [ Links ]
8. Houghton K, Malleson P, Cabral D, Petty R, Tucker L. Primary Sjögrens Syndrome in Children and Adolescents: Are Proposed Diagnostic Criteria Applicable? J Rheumatol 2005;32:2225-32. [ Links ]
9. Muscal E, DeGuzman M, Jung L, Flaitz C. Pediatric Sjogren Syndrome. Disponível em: http://emedicine.medscape.com/article/1008536-overview [ Links ]
10. Cedalia A. Sjögren Syndrome. In: Kliegman RM, Behrman RE, Jenson HB, Stanton BF. Nelson Textbook of Pediatrics. 18th Edition. Philadelphia: Saunders Editors; 2007:1028-9. [ Links ]
11. Tucker LB. Sjögren syndrome In: Cassidy JT. Petty RE, Laxer RM, Lindsley CB. Textbook of Pediatric Rheumatology. 6th edition. Philadelphia: Saunders Elsevier; 2011:548-65. [ Links ]
12. Panush R, Kramer N, Rosenstein E. Undifferentiated systemic rheumatic (connective tissue) diseases and overlap syndromes. Disponível em: http://www.uptodate.com/contents/undifferentiated-systemic-rheumatic-connective-tissue-diseases-and-overlap-syndromes [ Links ]
13. Cervera R, Khamashta MA, Hughes GR. Overlap syndromes. Ann Rheum Dis 1990; 49:947-8. [ Links ]
14. Hara T, Nagata M, Mizuno YU, Matsuo M, Ueda K, Recurrent parotid swelling in children: clinical features useful for differential diagnosis of Sjögrens syndrome. Acta Paediatr 1992; 81:547-9. [ Links ]
15. Aoki A, Ono S, Ueda A, Hagiwara T, Tsuji T, Misumi M, et al. Myositis in primary Sjögrens syndrome: clinical and pathological report. Mod Rheumatol 2003; 13:57-61. [ Links ]
16. Sjögren syndrome. Disponível em: http://www.mdconsult.com [ Links ]
17. Venables PJ. Management of patients presenting with Sjögren syndrome. Best Pract Res Clin Rheumatol 2006; 20:791-807. [ Links ]
Aida Silva e Sá

