










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.22 no.1 Porto mar. 2013
Síndrome de Hanhart – caso clínico
Hanhart syndrome – clinical report
Helena Rios1, Cátia Carnide2, Sofia Morais1, Miguel Branco2, Joana Mesquita1, Eulália Galhano2, Lina Ramos3
1 S. Neonatologia, Dep. Saúde da Mulher, CH e Universitário de Coimbra
2 S. Obstetrícia, Dep. Saúde da Mulher, CH e Universitário de Coimbra
3 S. Genética Médica, Dep. Pediátrico, CH e Universitário de Coimbra
RESUMO
Introdução: Em 1950 Hanhart descreveu três casos de aglossia e deformidades dos membros. A associação de malformações oromandibulares e dos membros é rara, verificando-se grande variabilidade fenotípica entre os casos descritos.
Caso Clínico: Gravidez com diagnóstico pré-natal ecográfico (24 semanas) de agenesia do pé direito, ausência do primeiro dedo do pé esquerdo e presença apenas da primeira falange do primeiro e quinto dedos da mão esquerda. Às 38 semanas de gestação nasce um recém-nascido, sexo masculino, com hipotonia generalizada e dificuldade respiratória com necessidade de manobras de reanimação. Ao exame físico destacava-se a presença de microretrognatia, microstomia e várias malformações ósseas a nível das mãos e pés.
Desde as primeiras horas de vida que apresentou quadro de apneias com dessaturações recorrentes vindo a falecer na decorrência de um desses episódios.
Discussão: A associação de microstomia, microretrognatia, hipoglossia, malformações dos membros e atingimento dos pares cranianos, permite-nos estabelecer o diagnóstico clínico de Síndrome de Hanhart. O seu diagnóstico nem sempre é fácil, dada a grande variabilidade fenotípica.
Palavras-chave: Síndrome de Hanhart, malformações oromandibulares e dos membros.
ABSTRACT
Introduction: In 1950, Hanhart described three cases of aglossia and limb malformations. The association of congenital oromandibular and limb malformations is rare and has a wide phenotypic variability among the cases described.
Case Report: Pregnancy with prenatal ultrasound diagnosis (24 weeks) of agenesis of the right foot, absence of the first finger of the left foot and presence of only the first phalange of the first and fifth fingers of the left hand. At 38 weeks of gestation, a male newborn was born with generalized hypotonia and respiratory distress requiring resuscitation. Physical examination showed microretrognathia, microstomy and several bone malformations of the hands and feet.
From the first hours of life he presented progressive apneas with dessaturation, dying as result of one of those episodes.
Discussion: The association of microstomy, microretrognathia, hypoglossia, limb malformations and signs suggestive of cranial nerve compromise, allows us to establish the clinical diagnosis of Hanhart Syndrome. The diagnosis is not always easy, given the wide phenotypic variability.
Keywords: Hanhart syndrome, oromandibular and limb malformations.
INTRODUÇÃO
A associação de malformações oromandibulares e dos membros é rara, tendo sido descrita pela primeira vez por Kettner (1907)(1). Posteriormente Rosenthal (1932) descreveu uma menina de três anos com microglossia, micrognatia, fenda labial, ausência de incisivos inferiores e deformidades das mãos e pés(2). Em 1950 Hanhart descreveu três casos de aglossia e deformidades dos membros(3). Desde então, há descrições na literatura de raros casos, verificando-se grande variabilidade fenotípica entre eles.
CASO CLÍNICO
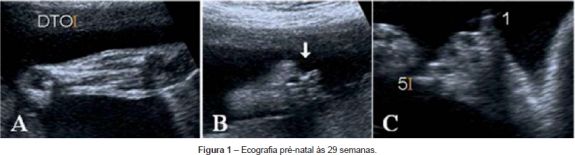
Gravidez vigiada sem intercorrências até às 24 semanas de gestação, altura em que é feito o diagnóstico pré-natal ecográfico de malformações dos membros. Às 29 semanas foi referenciada à nossa maternidade, onde repete ecografia que mostra: agenesia do pé direito (Figura 1A), ausência do primeiro dedo do pé esquerdo (Figura 1B), identificando-se apenas a primeira falange do primeiro e quinto dedos da mão esquerda (Figura 1C); a restante avaliação ecográfica não demonstrou outras malformações. O cariótipo fetal era normal (46,XY), assim como o ecocardiograma fetal. Não tinha antecedentes familiares relevantes, nomeadamente malformações congénitas ou consanguinidade e não tinha ocorrido exposição a agentes teratogénicos durante a gestação.
O recém-nascido, do sexo masculino, nasceu às 38 semanas, com hipotonia generalizada e dificuldade respiratória, necessitando de medidas de reanimação. Por apresentar respiração irregular e necessidade de O2 suplementar foi internado na Unidade de Cuidados Intensivos Neonatais.
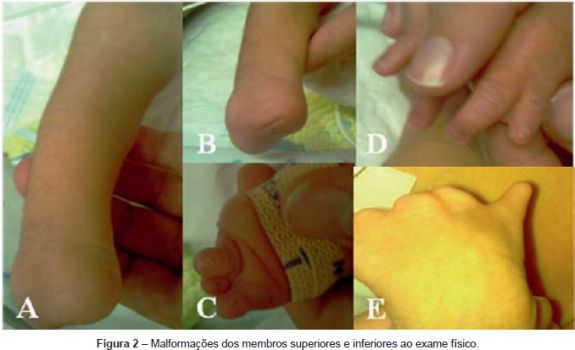
Ao exame físico apresentava as seguintes alterações: microretrognatia, rigidez mandibular, hipoglossia e microstomia; encurtamento do membro inferior direito com ausência do pé do mesmo lado (Figuras 2A, 2B e 3A), ausência de um dedo do pé esquerdo com encavalgamento e sindactilia dos restantes dedos (Figura 2C); sindactilia entre o segundo e terceiro dedos da mão direita (Figura 2D) e ausência das falanges distais e sindactilia dos dedos da mão esquerda (Figura 2E e 3B); cifoescoliose (Figura 3C). A auscultação pulmonar revelou diminuição bilateral do murmúrio vesicular e ao exame neurológico apresentava fraca mobilidade espontânea, face inexpressiva, gemido, ausência dos reflexos de sucção e deglutição e hipotonia generalizada.
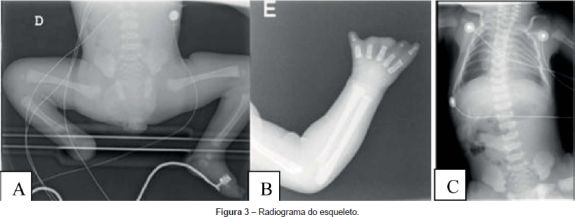
O radiograma do esqueleto confirmou as anomalias descritas anteriormente (Figura 3).
Na restante investigação efetuada não foram identificadas alterações significativas: ecografias cardíaca e transfontanelar, diagnóstico precoce e MLPA (Multiplex ligation-dependent probe amplification) das regiões subteloméricas.
Desde o primeiro dia de vida que apresentou apneias com dessaturações recorrentes difíceis de recuperar apesar da aspiração contínua de secreções, necessitando de ventilação não invasiva a partir do segundo dia. Ao sexto dia teve um episódio de apneia, do qual não recuperou com as medidas de reanimação.
DISCUSSÃO
A associação de microstomia, microretrognatia, hipoglossia, adactilia /hipodactilia /agenesia congénita de mãos e pés e presença de sinais neurológicos sugestivos de atingimento dos pares cranianos, permite-nos estabelecer o diagnóstico clínico de Síndrome de Hanhart.
A Síndrome de Hanhart é uma doença rara (prevalência de 1:500.000 nascimentos) (4,5). Alterações que ocorrem entre a 4ª e 8ª semana de embriogénese condicionarão as malformações que caracterizam esta síndrome, cuja etiologia é desconhecida(5). Alguns autores apontam a possibilidade de ter uma base genética, secundária a mutações esporádicas(5-7). Outros salientam a importância de fatores ambientais como: fatores intrauterinos (bandas fibróticas, alterações vasculares), fármacos teratogénicos e biópsia das vilosidades coriónicas antes das 10 semanas de gestação(4,5,7,8).
O diagnóstico da Síndrome de Hanhart é clínico e radiológico(6). A presença de micrognatia e/ou aglossia/microglossia e de alterações do desenvolvimento dos segmentos distais dos membros são essenciais para o diagnóstico(5,7). Estas alterações dos membros são geralmente assimétricas e com atingimento mais frequente dos membros superiores. No nosso caso, o recém-nascido apresentava micrognatia, hipoglossia e microstomia com malformações assimétricas dos membros superiores e inferiores (agenesia de um pé, sindactilia, adactilia, hipodactilia).
No entanto, para além destas são descritas outras malformações que contribuem para a grande variabilidade fenotípica, e consequente dificuldade do diagnóstico: anquilose temporo-mandibular, fenda do palato, alterações dos dentes (hipodontia, amelogenese imperfeita, atraso da erupção dentária, problemas de implantação dentária), hipoplasia ungueal e parésia congénita de pares cranianos(4,5,7). Estão também descritos casos associados a ânus imperfurado, fusão espleno-gonadal, hipoplasia pulmonar(5).
As principais complicações traduzem-se nas limitações físicas inerentes às malformações dos membros, dificuldades alimentares e problemas respiratórios(5). Apresentam ainda dificuldades de audição e linguagem, no entanto sem alterações na função cognitiva(4,5,7).
No período neonatal a existência de compromisso da alimentação e insuficiência respiratória (consequentes das alterações oromandibulares e compromisso dos pares cranianos) poderá levar à morte destas crianças(5). No nosso caso, o recém-nascido evoluiu para uma insuficiência respiratória, decorrente de um quadro de apneias recorrentes.
Após o período neonatal, o tratamento reside na colocação de próteses nos membros e na cirurgia de reconstrução oromandibular, a qual permite a resolução dos problemas de deglutição(5). Nalguns casos é necessário a colocação de gastrostomia para uma adequada nutrição(6).
Os autores consideram que a apresentação deste caso é importante dada a raridade do mesmo. Ao conhecer as principais alterações ao exame físico que o caracterizam é possível estabelecer o seu diagnóstico clínico e assim oferecer um mais adequado apoio pré e perinatal.
BIBLIOGRAFIA
1. Kettner D. Kongenitaler Zungendefekt. Deutsche Medizinische Wochenschrift 1907; 33:532. [ Links ]
2. Rosenthal R. Aglossia congenita: report a case of the condition combined with other congenital malformations. Am J Dis Child 1932; 44:383-9. [ Links ]
3. Hanhart E. Uber die Kombination von peromelie mit mikrognathie: ein neues syndrome bein Menschen, entsprechend der akroteriasis congenital von wreidt und mohr, beim rinde. Arch Julius Klaus Stift Vererbungsforsch 1950; 25:531-40. [ Links ]
4. Castillo ST, Rojas JZ, Monasterio LA. Síndrome de Hanhart. Rev Child Ped 1985; 56:180-3. [ Links ]
5. Estrada RC, Rivas RG, Marcos RA, Romero AB, Lezcano AR. Síndrome de Hanhart (Síndrome de aglossia-adactilia). A propósito de dos casos. An Esp Pediatr 1990; 33:465-8. [ Links ]
6. Postai GV, Freitas RS, Roça GB, Menacho AM, Souza J, Alonso N, Passos-Bueno MR. Síndrome de Hanhart: relato de casos. Arquivos Catarinenses de Medicina 2009; 38:278-80. [ Links ]
7. Nevin NC, Burrows D, Allen G, Kernohan DC. Aglossia-adactylia syndrome. J Med Genet 1975; 12:89-93. [ Links ]
8. Firth HV, Boyd PA, Chamberlain PF, Mackenzie IZ, Morris-Kay GM, Huson SM. Analysis of limb reduction defects in babies exposed to chorionic villus sampling. Lancet 1994; 343:1069-71. [ Links ]
Helena Rios

