










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.22 no.3 Porto set. 2013
CASO CLÍNICO / CASE REPORT
Deleção intersticial 8p23.1-8p23.2 – caso clínico de diagnóstico pós–natal
Interstitial deletion 8p23.1-8p23.2 – a case report of a postnatal diagnosis
Vânia FerreiraI; Raquel MacielI; João CasanovaI; Sílvia ÁlvaresII; Natália Oliva TelesIII; Manuela Mota FreitasIII; Maria do Céu RodriguesI; Maria José MendesI
IS. Obstetrícia e Ginecologia, U. Diagnóstico Pré-Natal, Maternidade Júlio Dinis, CH Porto, 4050-371 Porto, Portugal. vania_ferreira@hotmail.com; raquelmaciel@gmail.com; joaomiguelcasanova@gmail.com; mcpprodrigues@gmail.com; mjosemendes688@gmail.com
IIS. Cardiologia Pediátrica, CH Porto, 4099-001 Porto, Portugal. silvia.c.alvares@gmail.com
IIIDep. Genética, U. Citogenética, Centro de Genética Médica Dr. Jacinto Magalhães, INSA, I.P., 4099-028 Porto, Portugal. manuela.freitas@chporto.min-saude.pt; natalia.teles@chporto.min-saude.pt
RESUMO
Introdução: A deleção cromossómica 8p23 apresenta um espectro clínico variável que se deve à extensão da deleção ou da região do ponto de quebra do material genético.
Caso clínico: Grávida de 23 semanas de gestação referenciada ao Centro de Diagnóstico Pré Natal por bradiarritmia fetal. A ecocardiografia fetal revelou anel pulmonar estreito e forâmen oval grande. Parto eutócico às 40 semanas de gestação de um feto do sexo masculino e com peso abaixo do percentil 10. A ecocardiografia pós-natal revelou: defeito perimembranoso do septo ventricular, comunicação interauricular e estenose moderada da válvula pulmonar. Durante as consultas de seguimento verificaram-se características dismórficas, atraso de desenvolvimento e alterações do comportamento. Foram realizados estudos citogenéticos e de citogenética molecular; o cariótipo de ambos os progenitores não revelou alterações. O cariótipo final da criança foi definido como: 46,XY,del(8)(p23.1p23.2)dn.
Conclusão: As alterações cardíacas estão relacionadas com a haploinsuficiência do gene GATA4. A deleção desta região crítica está também associada a atraso mental ligeiro, alterações do comportamento e dismorfia facial ligeira, características presentes no espectro clínico do caso apresentado.
Palavras-chave: Aconselhamento genético, alterações cardíacas congénitas, análise citogenética, deleção 8p23.1, diagnóstico pré-natal, GATA-4.
ABSTRACT
Introduction: The features of an 8p23 deletion are likely to be a result of the loss of a number of different genes found in this region. The variable clinical features could be explained by the extent of the deletion or variation in the breakpoints.
Case report: A pregnant patient was referred for fetal echocardiography at 23 weeks gestation, because of fetal bradyarrhythmia, which showed a large foramen oval and mild pulmonary stenosis. A fetal male was born at 40 weeks, with a birth weight below the 10th centile. Postnatal echocardiography revealed: perimembranous ventricular septal defect, atrial septal defect and mild pulmonary valvar stenosis. During follow-up, dysmorphic features, development delay and behavioural issues were noticed. Cytogenetic and molecular cytogenetic analysis were performed. Parental chromosomes were normal, so the childs final karyotype was defined as 46,XY,del(8)(p23.1p23.2)dn.
Conclusion: Congenital heart defects are related with haploinsufficiency of gene GATA4. The deletion of this critical region is associated with mild mental retardation, behavioral problems and mild facial dysmorphy.
Keywords: Congenital heart defect, cytogenetic analysis, 8p23.1 deletion syndrome, GATA-4, genetic counselling; prenatal diagnosis.
INTRODUÇÃO
A deleção cromossómica 8p23.1 está associada a um espectro de anomalias clínicas que incluem dismorfia facial, microcefalia, atraso do crescimento intra-uterino, alterações neuropsiquiátricas, défice intelectual, cardiopatias congênitas (defeitos do septo atrioventricular, defeitos septais atriais, defeitos do septo ventricular, estenose pulmonar, tetralogia de Fallot) e hérnia diafragmática congénita.(1-5)
Em estudos publicados, as deleções que envolvem esta alteração genética variam desde deleções terminais extensas, que são facilmente detetadas por análise cromossómica de rotina, a deleções intersticiais que são melhor identificadas usando técnicas mais específicas como a hibridização in situ por fluorescência – FISH (fluorescence in situ hibridization) ou técnicas de citogenética molecular como a hibridização genómica comparativa – CGH.(4,6) Há relatos de deleções intersticiais e terminais do braço curto do cromossoma 8 (mais frequentemente com quebras nas bandas 8p21 a 8p23), bem como deleções ocorrendo em associação com duplicações. O quadro clínico variável deve-se à extensão da deleção ou à localização da região do ponto de quebra.(2,4,6)
O facto da deleção 8p23.1 estar associada a uma incidência aumentada de malformações cardíacas, sugere que esta região contém um gene importante no desenvolvimento do coração. Nos estudos publicados, identificou-se um gene que se encontrava recorrentemente excluído nos casos de deleções 8p23.1 – o gene GATA4, que foi considerado como o responsável por defeitos cardíacos associados com este tipo de deleções.(1,3-8) Verificou-se que o gene GATA4 se situa na porção proximal da banda p23.1 e codifica um fator de transcrição (estrutura em dedo de zinco) que desempenha um papel essencial na morfogénese cardíaca, nomeadamente na regulação de genes críticos para a diferenciação e função miocárdica. O papel do GATA4 no desenvolvimento do coração é apoiado por estudos de modelos de ratos e de indivíduos com mutações no referido gene. Nos modelos de ratos, tem sido demonstrado que deleções do gene GATA4 causam defeitos cardíacos. Os estudos demonstraram que ratos homozigóticos para um alelo nulo GATA4 não conseguiam formar um tubo cardíaco central e este defeito era fatal, cerca do 7º dia ao 9º dia pós coito(6,9).
O gene GATA4 tem sido implicado na regulação de genes críticos para a diferenciação e função do miocárdio, incluindo a troponina C, a α miosina cardíaca de cadeia pesada e o factor natriurético cerebral, sugerindo que os defeitos neste gene podem ter inúmeras implicações no desenvolvimento cardíaco.(7,9) Um estudo recente sugere que um outro gene, SOX7 (expresso no coração e que funciona na mesma via que o GATA4) tem também um papel importante no desenvolvimento cardíaco. Este gene está localizado na porção distal do braço curto do cromossoma 8, e nos casos de deleção 8p23 a haploinsuficiência deste gene tem também sido implicada nas anomalias cardíacas.(6)
O défice intelectual é o achado mais frequentemente descrito (relacionado com o desenvolvimento mental) neste tipo de deleções.(2) Parece existir uma relação entre o tamanho da região suprimida no cromossoma 8 e o grau de comprometimento intelectual - deleções terminais mais distais à banda 23.1 estão associadas a um menor compromisso intelectual.(3,8) Foram descritos outros tipos de desenvolvimento atípico neste tipo de deleções, nomeadamente défices motores, atraso da linguagem e alterações do comportamento tais como hiperatividade, défice de atenção e agressividade.(2) As deleções do gene SOX7 também têm sido propostas como tendo um papel no atraso do desenvolvimento e, possivelmente, nas características dismórficas verificadas nos indivíduos com deleção 8p23.(4,6,8)
Vários estudos têm tentado encontrar uma região crítica para os problemas de comportamento associados com deleções 8p23 e propuseram um gene, TNKS (Tankyrase 1), como o responsável pelos problemas de comportamento e dificuldades de aprendizagem, uma vez que é expresso no cérebro em grande quantidade. O gene TNKS tem também sido sugerido como o responsável pelas hérnias diafragmáticas, como observado em alguns indivíduos com deleções 8p23.(4,6) O gene MCPH1 (microcefalina 1) tem sido implicado no autismo, microcefalia e atraso do desenvolvimento, e também está localizado na banda p23 do cromossoma 8.(10)
CASO CLÍNICO
Grávida de 21 anos, foi referenciada à consulta de diagnóstico pré-natal às 23 semanas de gestação por bradiarritmia fetal. Realizou ecocardiografia fetal que revelou ritmo sinusal, frequência cardíaca normal, foramen oval grande (Figura 1) e anel pulmonar estreito (Figura 2). Parto eutócico às 40 semanas de gestação de um feto do sexo masculino, com um peso de 2290g (< percentil 10 - gráficos de crescimento de Lubchenco) e índice de Apgar 9/10.
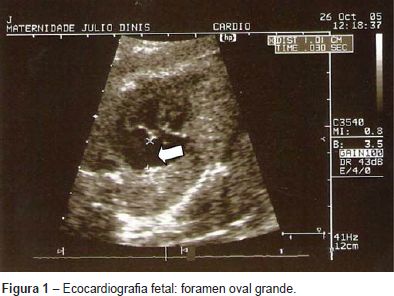
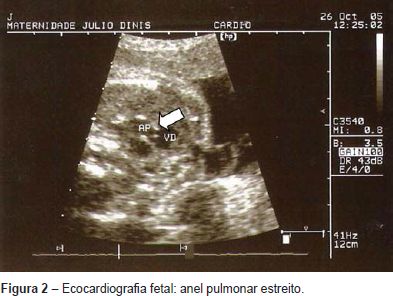
A ecocardiografia fetal pós-natal revelou uma comunicação interventricular perimembranosa (Figura 3a), comunicação interauricolar (Figura 3b) e estenose moderada da válvula pulmonar (Figura 4). Durante o acompanhamento, na consulta de cardiologia pediátrica, outras anomalias foram notadas, tais como: dismorfia facial, atraso do crescimento, pectus carinatum, atraso no desenvolvimento psico-motor e da linguagem (a criança sentou-se aos oito meses, disse as primeiras palavras aos 15 meses e começou a andar aos 18 meses), assim como alterações do comportamento (hiperatividade). A análise de citogenética molecular (FISH) para o cromossoma 22 foi normal e a análise cromossómica com bandas GTG de alta resolução revelou um cariótipo 46,XY,del(8)(p23.1). Foram realizadas outras técnicas de análise citogenética: 1) sonda de pintura cromossómica total (wcp) específica para o cromossoma 8 (whole chromosome painting probes, Cytocell) que demonstrou não estar envolvido outro cromossoma além do 8; 2) sondas subteloméricas para o cromossoma 8 cujo resultado não revelou alterações, tendo-se evidenciado que a deleção era intersticial. Os estudos genéticos dos progenitores foram normais, pelo que a deleção detetada no filho foi considerada de novo. Assim, o cariótipo final da criança foi definido como: 46,XY,del(8)(p23.1p23.2)dn.
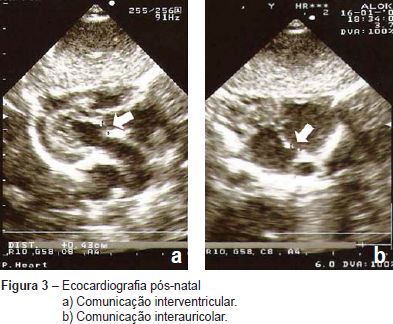
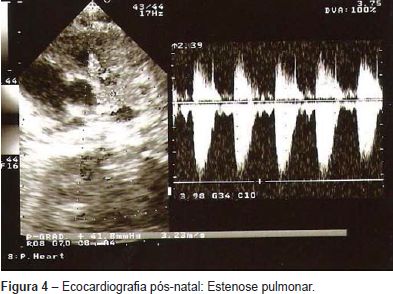
Aos cinco anos de idade a criança foi submetida a cirurgia corretiva cardíaca, que consistiu no encerramento do defeito do septo ventricular e da comunicação interauricular com um patch pericárdico e comissurotomia pulmonar. Atualmente, com oito anos de idade, está assintomático do ponto de vista cardíaco, apresentando um atraso psicomotor e um comportamento hiperativo.
DISCUSSÃO
Neste caso clínico descrevemos o fenótipo atípico de um indivíduo com deleção intersticial 8p23.1-8p23.2, sendo o primeiro caso descrito em Portugal. Vários autores têm sugerido que as deleções 8p podem ser mais frequentes do que o número relativamente baixo de casos descritos na literatura.(1-3) Essa realidade deve-se ao facto de, possivelmente, apenas crianças com atraso de desenvolvimento ou anomalias major serem referenciadas para estudos citogenéticos, enquanto aqueles com poucas manifestações ou com alterações subtis permanecerem sem diagnóstico.(2)
Há muitos relatos de deleções subteloméricas 8p. Os estudos iniciais, publicados no século XX, foram notificados como deleções terminais, no entanto, quando os mesmos indivíduos foram reavaliados por técnicas moleculares e citogenéticas mais avançadas (incluindo marcadores de microssatélites e análise por FISH), foram diagnosticadas deleções intersticiais em vez de terminais.(4)
A criança apresentada neste caso clínico apresenta achados clínicos comumente descritos em indivíduos com deleção 8p23, tais como: atraso do crescimento, dismorfia facial leve, malformações cardíacas e atraso do desenvolvimento. Os resultados de estudos de correlação genótipo-fenótipo de indivíduos com defeitos cardíacos congênitos destacaram a região cromossómica 8p23.1 como uma região crítica para a morfogénese cardíaca. Esta região contém o gene GATA4 que codifica um fator de transcrição que desempenha um papel fulcral no desenvolvimento do coração em seres humanos. As cardiopatias congénitas nas deleções deste tipo podem ser explicadas pela haploinsuficiência do gene GATA4(1,3-10).
A região crítica para o atraso mental ligeiro, alterações do comportamento e dismorfia facial também se localiza na banda p23.1 do cromossoma 8(4,6,10), e essas características estão presentes na criança do caso clínico apresentado.
Estes dados reforçam a importância de realizar uma cuidadosa análise citogenética/ molecular da região 8p23.1, no período pré e pós-natal, nos indivíduos que apresentam cardiopatias congénitas.
É fundamental correlacionar a análise clínica e citogenética com a finalidade de esclarecer a correlação genótipo/fenótipo, uma vez que isso nos permitirá uma maior acuidade no aconselhamento genético.
Os autores esperam que, com este caso clínico, tenham contribuído para um maior conhecimento científico destes tipos de deleções.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Clayes I, Holvoet M, Eyskens B, Adriaensens P, Gewillig M, Fryns JP, et al. A recognisable behavioural phenotype associated with terminal deletions of the short arm of chromosome 8. Am J Med Genet 1997; 74:515-20. [ Links ]
2. Gilmore L, Cuskelly M, Jobling A, Smith S. Deletion of 8p: a report of a child with normal intelligence. Dev Med Child Neurol 2001; 43:843-6. [ Links ]
3. Hutchinson R, Wilson M, Voullaire L. Distal 8p deletion (8p23.1-pter): A common deletion? J Med Genet 1992; 29:407-11. [ Links ]
4. Páez MT, Yamamoto T, Hayashi K, Yasuda T, Harada N, Matsumoto N, et al. Two patients with atypical intersticial deletions of 8p23.1: Mapping of phenotypical traits. Am J Med Genet Part A 2008; 146A:1158-65. [ Links ]
5. Wu BL, Schneider GH, Sabatino DE, Bozovic LZ, Cao B, Korf BR. Distal 8p deletion (8)(p23.1): An easily missed chromosomal abnormality that may be associated with congenital heart defect and mental retardation. Am J Med Genet 1996; 62:77-83. [ Links ]
6. Wat MJ, Shchelochkov OA, Holder AM, Breman AM, Dagli A, Bacino C, et al. Chromosome 8p23.1 deletions as a cause of complex congenital heart defects and diaphragmatic hernia. Am J Med Genet Part A 2009; 149A:1661-77. [ Links ]
7. Bhatia SN, Suri V, Bundy A, Krauss CM. Prenatal detection and mapping of a distal 8p delection associated with congenital heart disease. Prenat Diagn 1999; 19:863-7. [ Links ]
8. Digilio MC, Giannotti A, Marino B, Dallapicola B. Atrioventricular canal and 8p-syndrome. Am J Med Genet 1993; 47:437-8. [ Links ]
9. Molkentin JD, Kalvakolanu DV, Markham BE. Transcription factor GATA4 regulates cardiac muscle-specific expression of the alpha-myosin heavy-chain gene. Moll Cell Biol 1994; 14:4947-57. [ Links ]
10. Ozgen HM, van Daalen E, Bolton PF, Maloney VK, Huang S, Cresswell L, et al. Copy number changes of the microcephalin 1 gene (MCPH1) in patients with autism spectrum disorders. Clin Genet 2009; 76:348-56. [ Links ]
Vânia Gisela Costa Arcanjo Ferreira
Centro Hospitalar do Porto
Maternidade Júlio Dinis
Unidade de Diagnóstico Pré-Natal, Serviço de Obstetrícia
Largo da Maternidade
4050-371 Porto, Portugal
E-mail: vania_ferreira@hotmail.com
Recebido a 10.10.2012 | Aceite a 21.01.2013

