










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.23 no.1 Porto mar. 2014
CASOS CLÍNICOS / CASE REPORTS
Angioedema recorrente – caso clínico
Recurrent angioedema – a case report
Sandrina MartinsI; Miguel SalgadoI; Filipa RaposoI; Diana PintoII; Isabel MartinhoI; Rita AraújoI
IS. Pediatria, H Santa Luzia, ULS Alto Minho, 4901-858 Viana do Castelo, Portugal. E-mail: marsandrina@gmail.com; jmvs@sapo.pt; Filipa_raposo@hotmail.com; imartinhopipa@sapo.pt; arita_araujo@hotmail.com
IIS. Pediatria, CH Porto, 4099-001 Porto, Portugal. E-mail: disilvapinto@hotmail.com
RESUMO
Introdução: O Angioedema hereditário (AEH) é uma causa rara de angioedema recorrente, resultante de um defeito a nível do gene que codifica o inibidor do C1 esterase (C1-INH). O edema envolve predominantemente os tecidos da face, membros, trato gastrointestinal e área genital. O envolvimento da laringe, apesar de menos frequente, constitui a expressão clínica mais grave, sendo potencialmente fatal.
Caso clínico: Descreve-se o caso clínico de uma criança do sexo feminino de oito anos de idade referenciada à consulta de pediatria por episódios recorrentes de angioedema. O estudo efetuado revelou tratar-se de um caso de AEH.
Discussão: O diagnóstico, estabelecido com base no quadro clínico, estudo do complemento e história familiar, é de importância fundamental considerando que o AEH é potencialmente fatal e exige uma terapêutica específica.
Palavras-chave: Angioedema hereditário, C1-INH, criança, recorrência.
ABSTRACT
Introduction: Hereditary angioedema (HA) is a rare cause of recurrent angioedema caused by a default in the gene that encodes the C1 esterase inhibitor (C1-INH). The oedema involves predominantly the face, limbs and genital and gastrointestinal tract. The involvement of the larynx, although less frequent, is the most severe clinical expression of HA and is potentially fatal.
Case report: Clinical report of an eight-year-old female with multiple episodes of angioedema. The laboratory study confirmed the diagnosis of HA.
Discussion: HA diagnosis is established based on the clinical history, family history and complements testing. Its documentation is extremely important because it is potentially fatal and needs specific therapy.
Key-words: C1-INH, children, hereditary angioedema, recurrence.
INTRODUÇÃO
O termo angioedema define o aparecimento súbito e transitório de edema do tecido celular subcutâneo e camadas profundas da derme, pelo aumento da permeabilidade vascular(1-3). Pode ser acompanhado de sensação dolorosa ou de queimadura e, tipicamente não é pruriginoso(1,2), a menos que acompanhado de urticária(3). Envolve áreas bem delimitadas atingindo preferencialmente a face, lábios, língua, mãos, pés, trato gastrointestinal e genitais(1,3). O compromisso da laringe, hipofaringe e língua pode condicionar asfixia e morte se não for instituído tratamento atempado e adequado(1,3). As causas e mecanismos fisiopatológicos de angioedema são diversos existindo formas adquiridas e, menos comumente, hereditárias(2,3). A sua abordagem é diferente e nem todas respondem adequadamente à adrenalina e corticóides(2,3). É essencial estabelecer o diagnóstico etiológico de forma a evitar a sua recorrência, permitindo também estabelecer o plano terapêutico mais adequado. Os autores apresentam uma causa rara de angioedema recorrente.
CASO CLÍNICO
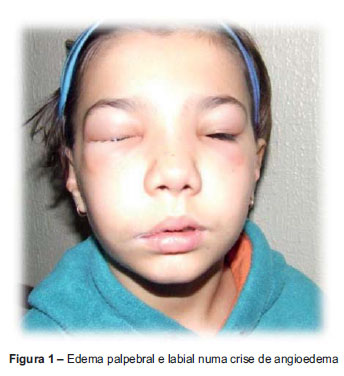
Criança de oito anos de idade, sexo feminino e raça caucasiana. Apresentava, episódios esporádicos de edema da face, mãos e/ou pés, desde os quatro anos de idade, não pruriginosos ou dolorosos, sem outra sintomatologia acompanhante. As queixas regrediam espontaneamente ou com terapêutica (anti-histamínico e/ou corticóide) em dois a três dias. Um aumento progressivo do número de crises, desde os seis anos de idade, motivou múltiplas vindas ao serviço de urgência (SU) e evicção escolar, sendo orientada para a consulta de Pediatria. O pai também apresentava, desde há três anos, edema ocasional dos genitais, pés e/ou face que resolviam espontaneamente. Sem outros antecedentes pessoais ou familiares relevantes. Perante o quadro clínico e a história familiar colocou-se a hipótese diagnóstica de angioedema hereditário (AEH), confirmada pela deteção de níveis baixos de C4 (4,1 mg/dl; pai: 4,5 mg/dl; N: 12-40) e C1-INH (7 mg/dl; pai: 9 mg/dl; N: 26-39) na criança e no pai. Os valores das frações C1q e C3 foram normais. Com o diagnóstico definitivo de AEH tipo I, foram dadas indicações sobre os procedimentos a seguir na crise aguda ou face a procedimentos endoscópicos, estomatológicos ou cirúrgicos (Quadro I), tendo sido prescrito adrenalina autoinjectável.
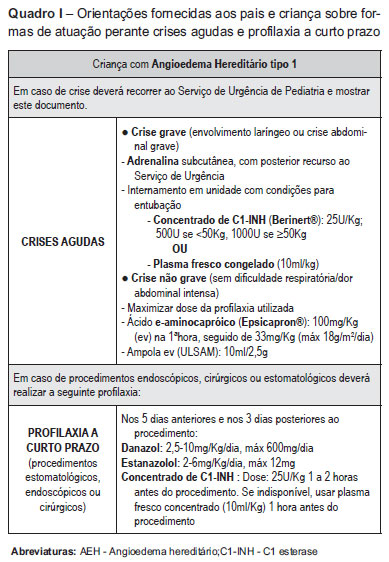
Em Novembro de 2010, na altura com sete anos e dois meses de idade, recorreu ao SU por quadro de edema palpebral bilateral e labial, duro e não doloroso (Figura 1), e dor abdominal periumbilical tipo cólica. Negava náuseas, vómitos, diarreia ou sintomas respiratórios. A sintomatologia tinha começado após traumatismo minor. Efetuou terapêutica com ácido e-aminocapróico e analgesia em regime de internamento tendo alta ap ós dois dias, assintomática. Atendendo a que nesta altura apresentava crises com periodicidade mensal, iniciou profilaxia a longo prazo com o agente anti-fibrinol ítico, sem recorrência posterior de episódios de angioedema.
DISCUSSÃO
O AEH, descrito pela primeira vez em 1888 por Osler(4,7), é uma causa rara de angioedema recorrente, com uma prevalência estimada de 1:50.000(4,5,9). É causado por um defeito a nível do gene que codifica o inibidor do C1 esterase (C1-INH)(1,2,4,5,9) localizado no cromossoma 11q(1,5,7-9,12), que condiciona um aumento da atividade da calicreína com vasodilatação e aumento da permeabilidade vascular (7-9,12).
Esta patologia caracteriza-se, como no caso clínico apresentado, por episódios recorrentes de angioedema, tipicamente não associados a urticária ou prurido(1-3,5-10,12,13), que envolvem predominantemente os tecidos da face, membros (particularmente as regiões distais), trato gastrointestinal e área genital(1,2,4-6,9,12). As crises abdominais podem apresentar-se como dor tipo cólica de forma isolada, como no caso apresentado, ou integrar-se em quadros mais graves com diarreia, vómitos(1,3-5,8) ou dor abdominal contínua e grave mimetizando frequentemente quadros de abdómen agudo(4,5,7,13). Formas raras de apresentação são: tosse e dor pleurítica associada a derrame pleural, convulsões e hemiparesia transitórias por edema cerebral e sintomas urinários(1,5,12). O envolvimento da laringe, apesar de menos frequente, constitui a expressão clínica mais grave da doença, sendo a principal causa de morte nestes doentes(1,2,4,5,7-9,12,13). Habitualmente os sintomas evoluem ao longo de 24 horas podendo ir acometendo diferentes áreas anatómicas, regredindo posteriormente em dois a cinco dias(1,3-5,7,9,12), com período intercrítico variável(12). No episódio que condicionou internamento e terapêutica com anti-fibrinolítico havia história de um traumatismo minor prévio, um dos desencadeantes de crises de AEH mais frequentemente relatado na literatura(1,4,5,8,9,12,13). Podem ainda ser predisponentes procedimentos estomatológicos ou cirúrgicos(1,4,5,13) , stress(1,4,8,9,12,13), infeções(1,8,9,12), fármacos (inibidores da enzima de conversão de angiotensina e anticoncecionais orais)(1,12) ou exposição ao frio(5), embora a maioria das crises seja espontânea não se identificando fator precipitante(3,7,8,12). A sintomatologia inicia-se geralmente na primeira ou segunda décadas de vida(5,12) e persiste durante toda a vida, sendo a evolução da doença imprevisível e existindo uma grande variabilidade de expressão fenotípica no próprio indivíduo ao longo do tempo ou em indivíduos da mesma família(4,5,7). O diagnóstico é estabelecido com base no quadro clínico, estudo do complemento e história familiar, embora 25% dos casos representem mutações de novo(3-5,7-9,12).
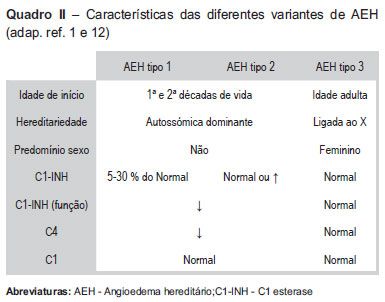
Existem duas principais formas de AEH transmitidas de forma autossómica dominante (4 5,8,9,13) de penetrância incompleta (11,12). O AEH tipo I, responsável por cerca de 85% dos casos(2,4-8) caracteriza-se, como no caso clínico apresentado, por um defeito quantitativo e qualitativo de C1-INH(2,4,5-9,12,13). No AEH tipo II existe um défice exclusivamente funcional da proteína(2,4,5-9,12,13) com um doseamento do antigénio normal ou por vezes aumentado(5, 6, 8, 9). Os níveis de C4 estão tipicamente diminuídos nos dois tipos e as frações de C1q e C3 normais(1,7). O doseamento de C4 representa, inclusivamente, um bom método de rastreio embora, em 1 a 2% dos casos, os seus níveis sejam normais no período intercrise(4,6-8,12). Do ponto de vista clínico, estas duas formas são indistinguíveis(4). Mais recentemente foi descrito o AEH tipo III, frequentemente associado a uma mutação no gene do fator XII da coagulação, em que os níveis e função do C1-INH são normais(4,5,6,8,9) (Quadro II).
O diagnóstico de angioedema hereditário é de extrema import ância tendo em conta que é potencialmente fatal e necessita de terapêutica específica, não tendo os anti-histamínicos e corticóides qualquer benefício no tratamento desta entidade(1,4,5,8-11). A adrenalina pode proporcionar um alívio transitório face ao seu papel vasoconstritor que impede a transudação de plasma(4,7,8,10,12), pelo que os doentes devem possuir adrenalina autoinjectável que deve ser administrado em caso de crises graves, sem dispensar a ida ao serviço de urgência. Atualmente, o tratamento de eleição na fase aguda, particularmente quando há envolvimento laríngeo ou crises abdominais graves(1), consiste na administra ção do concentrado de C1-INH que corrige diretamente o défice proteico(1,4,10,12). Se indisponível, o uso de plasma fresco congelado (PFC) constitui uma alternativa embora esteja associado a um risco de exacerba ção paradoxal da clínica por veicular outros substratos da cascata de coagulação(1,4,7,10,13). Ainda na fase aguda, e no caso de crises menos severas, o uso de antifibrinol íticos (ácido e-aminocapróico e ácido tranexámico) ou androgénios modificados (danazol ou estanozolol) em doses elevadas têm demonstrado eficácia(1), (8,12). O tratamento de suporte não deve ser descurado e é fundamental ter em conta que as modalidades terapêuticas descritas duram cerca de 30 minutos a uma hora a surtir efeito. Assim, no caso de surgirem sinais de obstru ção da via aérea superior como estridor, disfonia ou sialorreia, a entubação endotraqueal deve ser prontamente executada(3,10,11). A analgesia, no caso de dor abdominal, é fundamental(1,3,10,12) e considerando a transudação de fluidos e o risco de hipovolemia pode haver necessidade de reposi ção do volume intravascular de forma a evitar choque hipovolémic (1,10,11).
Para além da terapêutica da fase aguda, a profilaxia a longo prazo deve ser ponderada nos casos em que existe pelo menos uma crise grave / potencialmente fatal ou nos casos em que as crises são muito frequentes – uma ou mais crises mensais, condicionando uma incapacidade mensal superior a cinco dias(1,4,7,8,11). No caso clínico apresentado, foi iniciada terapêutica profilática dada a frequência das crises. Dispomos atualmente de três opções: antifibrinolíticos(4,8,11-13) androgénios modificados(4,8,11-13) e administração de concentrado de C1-INH a cada quatro a sete dias(1,7,8,11,12). A administração de C1-INH é uma terapêutica dispendiosa e incómoda. Deve ser reservada para os casos em que as outras duas alternativas não controlam a doença ou no caso de haver contraindicações ao seu uso(11,12). Os androgénios modificados, apesar de aparentemente mais eficazes que os antifibrin olíticos(1,4,8,11-13), são reservados para os casos refratários, devido aos seus efeitos laterais. Em idade pediátrica, os antifibrinolíticos constituem o tratamento de primeira linha(1,4,7,8,11-13). Atualmente em Portugal apenas é comercializado o ácido-o-aminocaproico apesar do ácido-tranexámico estar aparentemente associado a melhores resultados e melhor tolerância(7,8,11). Os efeitos laterais desta terapêutica incluem alterações gastrointestinais, cefaleias, tonturas, mialgias, rabdomiólise(4,8), trombose vascular(1,4,7), hipotensão postural(7,8), insuficiência renal(7) e lesão da retina(7,9).
Na profilaxia a curto prazo, indicada para procedimentos estomatológicos, endoscópicos ou cirúrgicos, existem como opções: o concentrado de C1-INH ou PFC administrados uma a seis horas antes(1,4,7,8,13) ou os androgénios modificados(4,7,8,13) a administrar desde cinco dias antes até três a cinco dias depois do procedimento(1,4,8,11,12). Esta última opção, além de ser menos dispendiosa tem-se mostrado segura e eficaz, embora o uso de C1-INH tenha indicação absoluta em casos de procedimentos cirúrgicos major, procedimentos minor em doentes com maior gravidade clínica, procedimentos não programados ou contraindicação à administração de androgénios modificados(7,8).
O uso de diversos fármacos, já aprovados para tratamento em adultos, com mecanismos de ação mais diretos e formas de administração mais cómodas, pode vir a ser o futuro do tratamento no AEH da população pediátrica. Entre eles destacam-se o C1-INH recombinante, um antagonista dos recetores B2 da bradicinina (Icatibant) e um inibidor da calicreína plasmática (ecallantide)(4,8,9).
A instituição de antifibrinolítico nesta criança foi acompanhada de boa resposta clínica tanto na crise aguda como a longo prazo, refletindo-se numa franca diminuição do números de crises, sem efeitos laterais importantes.
É de importância primordial nestes casos fornecer esclarecimentos sobre a doença e seus riscos e ceder informação escrita que acompanhe sempre o doente, onde conste o tratamento a efetuar em caso de crise (Quadro I).
Face à baixa prevalência da doença, o diagnóstico de angioedema hereditário implica um elevado índice de suspeita, nomeadamente em casos de angioedema recorrente sem urticária associada(3). Num estudo efetuado em 2005, o intervalo entre o início da sintomatologia e o diagnóstico foi de 10 anos(4). No caso relatado este intervalo foi de cerca de quatro anos e o diagnóstico foi sugerido pela presença de história familiar positiva, com confirmação da doença no pai. Salienta-se, pois, a importância do rastreio familiar para deteção precoce da doença e aconselhamento genético.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Martins P, Gaspar A, Pires G, Godinho N, Almeida M, Pinto J. Angioedema hereditário em idade pediátrica. Rev Port Imunoalergologia 2003; XI:410-20. [ Links ]
2. Hassan G, Khan G, Qureshi W, Ibrahim M. Angioedema: current concepts. JK Science 2005; 7:133-34. [ Links ]
3. Bingham C. An overview of angioedema: Clinical features, diagnosis, and management. Up to Date; versão 19.2. Disponível em: www.uptodate.com [ Links ]
4. Zuraw B. Hereditary angioedema. N Engl J Med 2008; 359:1027-36. [ Links ]
5. Atkinson J, Cicardi M, Sheffer A. Clinical manifestations and pathogenesis of hereditary angioedema. Up to Date; versão 19.2. Disponível em: www.uptodate.com [ Links ]
6. Atkinson J, Cicardi M, Sheffer A. Diagnosis of hereditary angioedema. Up to Date; versão 19.2. Disponível em:www.uptodate.com [ Links ]
7. Paiva M, Gaspar A, Loureiro V, Pinto P. Angioedema hereditário – caracterização de uma população pediátrica. Rev Port Imunoalergologia 2010; 18:157-74. [ Links ]
8. Bowen T, Cicardi M, Farkas H, Bork K, Longhurst HJ, Zuraw B, et al. 2010 international consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol 2010; 6: 24-36. [ Links ]
9. Banerji A. Current treatment of hereditary angioedema: an update on clinical studies. Allergy Asthma Proc 2010; 31:398-406. [ Links ]
10. Atkinson J, Cicardi M, Sheffer A. Treatment of acute attacks in hereditary angioedema. Up to Date; versão 19.2. Disponível em: www.uptodate.comLinks ]uptodate.com/">
11. Atkinson J, Cicardi M, Sheffer A. Prevention of attacks in hereditary angioedema. Up to Date; versão 19.2. Disponível em: www.uptodate.comLinks ]uptodate.com/">
12. Cadinha S, Castel-Branco M, Malheiro D, Lopes I. Protocolo de diagnóstico, tratamento e seguimento de doentes com angioedema hereditário. Rev Port Imunoalergologia 2005; 13:377-93. [ Links ]
13. Papamanthos M, Matiakis A, Tsirevelou P, Kolokotronis A, Skoulakis H. hereditary angioedema: three cases report, members of the same family. J Oral Maxillofac Res 2010; 1:1-7. [ Links ]
Endereço para correspondência
Sandrina Martins
Rua dos Ferradores, nº 64
4740-448 Forjães Esposende, Portugal
E-mail: marsandrina@gmail.com
Recebido a 20.01.2012 | 26.09.2013

