










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.24 no.1 Porto mar. 2015
QUAL O SEU DIAGNÓSTICO? / WHAT IS YOUR DIAGNOSIS?
Caso electroencefalográfico
Electroencephalographic case
Sofia AiresI,II, Joana CarvalhoI,III, Rui ChorãoI,IV
IServiço de Neurologia Pediátrica, CH Porto. 4099-001 Porto, Portugal. Email: sofiaires@gmail.com; ju_carvalho1@hotmail.com; rui.chorao@gmail.com
IIServiço de Pediatria, CH Tondela-Viseu. 3509-504 Viseu, Portugal.
IIIServiço de Pediatria, CH Trás-os-Montes e Alto Douro. 5000-508 Vila Real, Portugal.
IVServiço de Neurofisiologia, CH Porto. 4099-001 Porto, Portugal.
RESUMO
Introdução: O Síndrome de Panayiotopoulos é a segunda epilepsia focal idiopática infantil mais comum, com um pico de incidência aos 4-5 anos. A sua incidência é subestimada devido às manifestações clínicas atípicas, caracterizando-se por crises com predomínio de sintomas autonómicos.
Caso clínico: Descreve-se um caso clássico de Síndrome de Panayiotopoulos, com crises recorrentes, com boa resposta à terapêutica antiepiléptica e remissão após alguns anos.
Conclusão: Salienta-se a importância da suspeição clínica deste síndrome epiléptico e a utilidade do registo electroencefalográfico que inclua um período de sono. Na maioria dos casos o prognóstico é bom, apesar de ser necessário manter o tratamento por um período de tempo variável.
Palavras-chave: epilepsia, Síndrome Panayiotopoulos.
ABSTRACT
Introduction: Panayiotopoulos syndrome is the second commonest idiopathic focal childhood epilepsy, with a peak of age at onset 4-5 years. Its incidence is usually underestimated, because of the typical unusual clinical manifestations of the seizures with predominant autonomic changes.
Case report: We describe a classical case of Panayiotopoulos syndrome, with recurrent seizures, but good response to antiepileptic drug treatment and remission after a few years.
Conclusion: The importance of the clinical suspicion of this epileptic syndrome and the usefulness of an electroencephalographic recording including a period of sleep is underlined. In most diagnosed cases prognosis is good, although therapy is usually necessary for a variable period of time.
Keywords: epilepsy, Panayiotopoulos syndrome.
CASO CLÍNICO
Descreve-se o caso de uma criança do sexo masculino, de cinco anos de idade, filho de casal saudável, não consanguíneo. Dos antecedentes pessoais, hérnia inguinal esquerda (herniorrafia aos 15 meses) e hipospádias (corrigida cirurgicamente aos 8 anos).
Referenciado à consulta por episódios, o primeiro aos quatro anos de idade e recorrendo com intervalos de vários meses, sempre relacionados com o sono, em que despertava, vomitava, ficando muito pálido e apático, com desvio cefálico e ocular para a esquerda. Estes episódios tinham a duração de 2 a 4 minutos, com resolução espontânea, após o que adormecia novamente.
O exame neurológico não revelou alterações.
Realizou electroencefalograma (EEG) que revelou actividade paroxística focal, parieto-occipital direita e também frontal direita, com acentuação no sono (figuras 1 e 2).
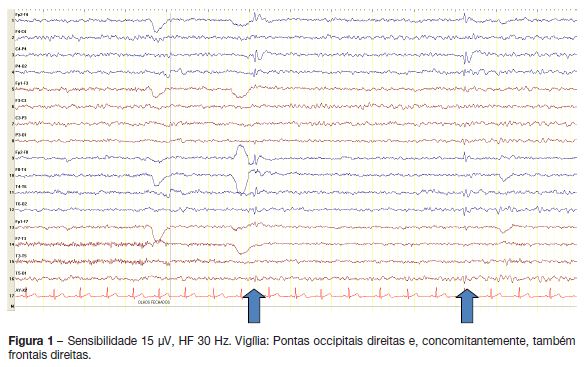
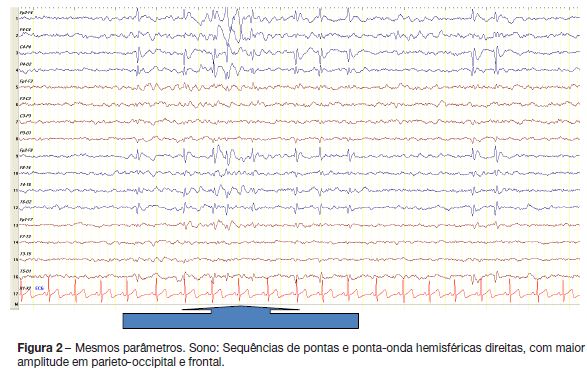
Qual é o seu diagnóstico?
Diagnóstico
Síndroma de Panayiotopoulos
EVOLUÇÃO
A imagem cerebral por ressonância magnética não mostrou alterações.
Foi medicado com carbamazepina na dose aproximada de 10 mg/kg/dia. Teve nova crise dois meses depois, com a mesma fenomenologia e evolução para crise hemiclónica esquerda. Foi aumentada dose de carbamazepina, ajustada ao aumento ponderal. Não voltou a ter crises até aos nove anos de idade, altura em que teve uma crise após adormecer no automóvel; parecia não ver, pelo menos em parte do campo visual, referia náuseas e tentava vomitar. Este episódio durou mais de 15 minutos, após o que ficou com cefaleia difusa e sonolência. Foi aumentada a dose do antiepiléptico, sem recorrência subsequente.
Por apresentar dificuldades escolares e quadro compatível com perturbação de hiperactividade e défice de atenção, foi medicado com metilfenidato, com benefício.
Por se manter sem crises e com menos alterações no EEG, sugerindo epilepsia não activa, não foi ajustada dose ao aumento ponderal e depois fez suspensão gradual, sem recorrência de crises e com alta da consulta aos 14 anos de idade.
DISCUSSÃO
A Síndroma de Panayiotopoulos é uma forma comum de epilepsia infantil idiopática reconhecida pela Liga Internacional contra a Epilepsia.1 É considerada uma manifestação mais precoce do espectro das epilepsias occipitais benignas ou autolimitadas da infância, sendo a síndroma de Gastaut uma manifestação mais tardia e com semiologia ictal diferente.2
Ocorre em crianças entre o 1 e os 14 anos, com um pico de incidência entre os 4-5 anos. A prevalência estimada de 13% em crianças com crises epilépticas apiréticas entre os três e os seis anos é provavelmente subestimada, dada a dificuldade diagnóstica, tendo em conta as características das crises.2,3
As crises manifestam-se essencialmente com sintomas autonómicos, sendo as náuseas e vómitos os sintomas mais comuns (em cerca de 80%). Podem surgir outras manifestações disautonómicas como palidez, midríase, alterações cardiorrespiratórias e termorreguladoras, incontinência de esfíncteres, hipersalivação e alterações da motilidade intestinal. O desvio ocular conjugado é muito frequente, podendo ocorrer em 60-80% dos casos. Em aproximadamente 20% dos casos há progressão para crise hemiclónica ou tónico-clónica generalizada, sendo também relativamente comum a perda de consciência não associada a convulsão (síncope ictal). Caracteristicamente, as crises são prolongadas, tipicamente com uma duração entre cinco a 10 minutos, mas em quase metade dos casos podendo durar mais de 30 minutos (estado de mal epiléptico disautonómico). A maioria das crises ocorre durante o sono, podendo ser também diurnas.2,4,5
O diagnóstico é clínico e suportado pelo EEG, que deve incluir necessariamente período de sono. O traçado interictal pode mostrar ondas abruptas ou pontas com diferentes localizações, podendo ser normal. Há um predomínio das alterações em occipital (aproximadamente 70% dos doentes em pelo menos um exame), não sendo, contudo, um pré-requisito para o diagnóstico. As anomalias são mais frequentes durante o sono e, na vigília, é característico o bloqueio dos paroxismos com a abertura dos olhos. Podem ocorrer paroxismos nas regiões frontais ou centro-temporais, concomitantes com os occipitais ou de forma isolada.2,4
O tratamento profi com antiepilépticos está indicado num grande número de doentes, pela recorrência das crises e tendo em conta que estas são frequentemente muito prolongadas. Os fármacos mais utilizados são a carbamazepina e o valproato.
O prognóstico é geralmente bom, com remissão das crises e sem repercussão neurológica. Um terço dos doentes têm apenas uma crise e apenas 10% têm mais que 10 crises ao longo de todo o período de epilepsia activa, mas existem casos de difícil controlo.4 O risco de desenvolver epilepsia na idade adulta é igual ao da população geral. Raramente, as crises autonómicas podem ser potencialmente fatais, tendo em conta o risco de paragem cardio-respiratória.3
Nesta síndroma, as características das crises (com predomínio dos fenómenos autonómicos e mimetizando outras situações) atrasam muitas vezes o diagnóstico.3 O reconhecimento desta situação pelos profissionais de saúde é crucial já que se trata de uma situação relativamente comum e geralmente com uma evolução favorável.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Engel Jr. A proposed diagnostic scheme for people with epileptic seizures and epilepsy: Report of the ILAE Task Force. Epileptic Disord 2001; 6: 121-4. [ Links ]
2. Covanis A, Ferrie, CD, Koutroumanidis M, Oguni H, Panayiotopoulos CP. Panayiotopoulos syndrome and Gastaur type idiopathic childhood occipital epilepsy. In: J Roger, M Bureau, Ch. Dravet, P. Genton, C.A. Tassinari & P. Wolf. Epileptic Syndromes in Infancy, Childhood and Adolescence (4th ed.). John Libbey Eurotext Ltd 2005, pp 227-53. [ Links ]
3. Covanis A. Panayiotopoulos Syndrome: a benign childhood autonomic epilepsy frequently imitating encephalitis, syncope, migraine, sleep disorder, or gastroenteritis. Pediatrics 2006;118;e1237. [ Links ]
4. Caraballo R, Cersósimo R, Fejerman N. Panayiotopoulos syndrome: A prospective study of 192 patients. Epilepsia 2007; 48(6): 1054-61. [ Links ]
5. Specchio N, Tivisano, DiCiommo, et al. Panayiotopoulos syndrome: A clinical, EEG, and neuropsychological study of 93 consecutive patients. Epilepsia 2010; 51(10):2098-107. [ Links ]
Endereço para correspondência Recebido a 07.01.2015 | Aceite a 12.02.2015
Rui Chorão
Serviço de Neurofisiologia
H. Santo António
Largo Prof. Abel Salazar 4099-001 Porto
E-mail: rui.chorao@gmail.com
