










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.25 no.2 Porto jun. 2016
CASOS CLÍNICOS / CASE REPORTS
A propósito de um caso de hipoglicemia...
A case of hypoglycemia…
Maria Miguel GomesI; Ângela PereiraI; Ariana AfonsoI; Helena SilvaI; Sofia MartinsII; Olinda MarquesII; Ana AntunesII
I S. de Pediatria, Hospital de Braga. 4710-243 Braga, Portugal. mariamgomes@hotmail.com; chelacbr@hotmail.com; ariana.afonso@gmail.com; tizleite@hotmail.com
II Grupo Endocrinológico Pediátrico, Hospital de Braga. 4710-243 Braga, Portugal. sofiacgam@gmail.com; opmarques@netcabo.pt; ana.antunes.p@gmail.com
RESUMO
Introdução: O hipopituitarismo é caracterizado por insufi- ciência da secreção hormonal hipófisária. A clínica é variável e depende da etiologia, evolução temporal e hormonas envolvidas.
Caso: Criança do sexo masculino com 2 anos, trazida à urgência por alteração súbita da consciência. No período neonatal apresentou quadro de hipoglicemia, trombocitopenia, icterícia e sépsis sem agente identificado. Objetivou-se crescimento regular no P10-25, desenvolvimento psicomotor adequado e estra- bismo divergente. Ao exame objectivo apresentava-se subfebril e com Escala de Coma de Glasgow 10. Foi constatada hipo- glicemia grave (26mg/dL) sendo realizado de imediato estudo endocrinológico e metabólico que mostrou cortisol baixo e défices de ACTH e GH; posteriormente foi confirmado défice de TSH. Iniciou terapêutica de substituição com hidrocortisona e levotiroxina. A neuroimagem mostrou alterações estruturais com hipoplasia da neuro-hipófise.
Conclusão: Este diagnóstico raro exige elevado grau de suspeição. A ocorrência progressiva dos défices hormonais obriga à avaliação clínica e laboratorial regular.
Palavras-chave: Hipopituitarismo; Hipoglicemia; Criança.
ABSTRACT
Introduction: Hypopituitarism is characterized by failure of the pituitary hormone secretion. Clinical presentation is variable and depends on the etiology, evolution and hormonal deficits.
Case: A 2 year-old male child was brought to the emergency room due to sudden change of consciousness. In the neonatal period, he presented with hypoglycemia, thrombocytopenia, jaundice and sepsis with no identified agent. The child showed regular growth (10-25 percentile), adequate psychomotor development and divergent strabismus. The patient was subfebrile with 10 Glasgow Coma Scale. Severe hypoglycemia (26mg/dL) was observed, and it endocrine and metabolic tests were immediately performed that showed low levels of cortisol, and ACTH, GH and TSH deficits. Replacement therapy with hydrocortisone and levothyroxine was started. Neuroimaging showed structural changes with adenohypophysis hypoplasia.
Conclusion: This rare diagnosis demands high degree of suspicion. The progressive development of hormonal deficits requires regular clinical and laboratory evaluations.
Keywords: Hypopituitarism; Hypoglycemia; Children.
INTRODUÇÃO
A díade hipotálamo-hipófise é dos componentes mais complexos do sistema endócrino.1-3 É fundamental na coordenação da resposta endócrina, estabelecendo relações de controlo mútuo sobre a maioria das glândulas endócrinas.1-3 As inter-relações estabelecidas entre hipotálamo-hipófise-glândulas endócrinas periféricas são exemplos de regulação por retrocontrolo.1-3 A perda destes normais mecanismos de regulação desencadeiam sinais e sintomas que, associados a doseamentos hormonais oportunos, podem dar o diagnóstico de patologias raras.1-3
A hipófise é composta por duas estruturas funcionalmente distintas que diferem na origem embriológica e anatómica: 1) adeno-hipófise/ pituitária anterior produtora de TSH (thyroid-stimulating hormone), FSH (follicle-stimulating hormone), LH (luteinizing hormone), GH (growth hormone), ACTH (adrenocorti- cotropic hormone), e prolactina; 2) neuro-hipófise/ pituitária posterior armazenadora de vasopressina e oxitocina.1-3
O hipopituitarismo é uma patologia rara, caracterizada por insuficiência parcial ou completa da secreção hormonal hipofisária.4 Pode ter origem em anomalias anatómicas (congénitas ou adquiridas) na hipófise, infundíbulo ou hipotálamo ou em mutações genéticas, assim como traumatismos, tumores ou doenças infiltrativas.4,5 A apresentação clínica é variável e depende de etiologia, evolução temporal e défices hormonais envolvidos.5
CASO CLINICO
Criança do sexo masculino com dois anos de idade. Antecedentes familiares irrelevantes e sem história de consanguinidade parental. Gestação de termo, com ecografias fetais descritas como normais e serologias maternas do terceiro trimestre sem evidência de infecção activa. Mãe sem hábitos tabágicos, etílicos ou consumo de outras drogas. Parto noutra instituição, por cesariana devido a apresentação pélvica, com índice de Apgar no 1º e 5º minuto de 7/9 e somatometria ao nascimento adequada à idade gestacional (peso 3145g no P25, comprimento 47,0cm no P25 e perímetro cefálico 36,0cm no P50). Rastreio de doenças endócrino-metabólicas normal em D5 e repetido em D20. Internamento no período neonatal por: hipoglicemias de D1 a D3, com resolução após aumento do aporte de glicose; trombocitopenia com necessidade de suporte transfusional em D2 (30000/uL plaquetas) e normalização em D14; icterícia com necessidade de fototerapia de D1 a D7 e D9 a D10 e icterícia colestática em D10 tratada com ácido ursodesoxicólico e vitaminas A, D, E e K até D15; sépsis sem agente identificado, tendo cumprido 10 dias de vancomicina e amicacina.
Evolução do crescimento regular no P10-25. Desenvolvimento psicomotor adequado, apresentando estrabismo diver- gente.
Previamente já tinha realizado ecografia transfontanelar aos 3 meses cujo resultado mostrou Cornos frontais dos ventrículos laterais com forma globosa e mais afastados que o comum. Aos 19 meses realizou Ressonância Magnética Nuclear (RMN) cerebral que revelou: Braquicefalia, incipiente hidrocefalia externa possível, heteropias nodulares periventriculares temporo-occipitais bilaterais e da coroa radiada esquerda.
Recorreu à urgência do Hospital de Braga por alteração do estado de consciência de início súbito. Encontrava-se sub-febril desde o dia anterior à admissão com tosse irritativa e obstrução nasal. Negava história prévia de traumatismo ou acesso a fár- macos/ tóxicos.
Frequentava infantário desde há 2 meses. Não tinha qualquer intercorrência infecciosa prévia.
Ao exame objectivo na admissão apresentava temperatura axilar de 37,5°C, frequência cardíaca 120 batimentos por minuto, tensão arterial 91/53 mmHg (sistólica P75 e diastólica P50). Prostrado e pouco reactivo, escala de coma de Glasgow 10 (O3 V2 M5), palidez cutânea e má perfusão periférica, sem sinais de dificuldade respiratória e sem sinais meníngeos. Auscultação cardíaca sem alterações. Auscultação pulmonar com murmúrio vesicular audível bilateralmente, com crepitações, tempo expiratório prolongado e raros sibilos. Abdómen mole e depressível, sem massas ou organomegalias palpáveis, sem sinais de irrita- ção peritoneal. Testículos palpáveis e pénis de características normais para a idade.
Realizou inicialmente pesquisa de glicemia capilar que não foi doseável e gasimetria venosa: pH 7,389, pCO 32,4 mmHg e HCO319,1 mmol/L, lactatos 1,76 mmol/L, glicose 26 mg/dL.
Foi administrado bólus de soro glicosado a 10% e iniciou fluidoterapia endovenosa, tendo-se verificado correção da gli- cemia e normalização progressiva do estado de consciência.
Foi colhido estudo analítico (Quadro 1) onde se confirmou hipoglicemia severa, presença de linfócitos atípicos (10%) e trombocitopenia.
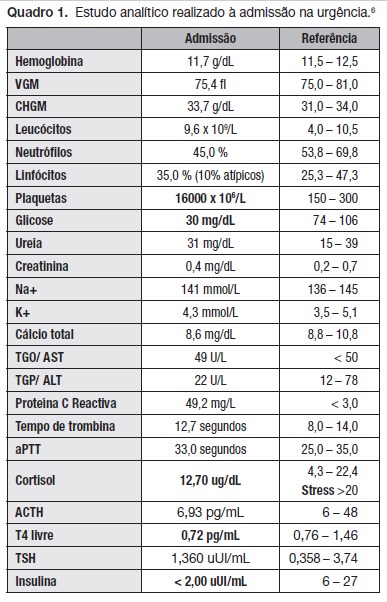
Perante hipoglicemia foi realizado de imediato estudo endocrinológico e metabólico que mostrou níveis de cortisol baixos para situação de stress. Os níveis de insulina estavam apropriadamente indoseáveis pelo que se pode excluir uma situação de hipoglicemia hiperinsulinémica.
A análise urinária, pesquisa de drogas de abuso, benzodiazepinas e etanol foi negativa.
A radiografia torácica, ecografia abdominal e tomografia computorizada cerebral foram descritas como normais.
Ficou internado para vigilância e investigação.
Manteve-se sempre apirético, hemodinamicamente estável e com perfil glicémico pré e pós-prandial normal.
Foi isolado vírus sincicial respiratório nas secreções.
A pesquisa de marcadores tumorais e substâncias redutoras na urina foram negativas.
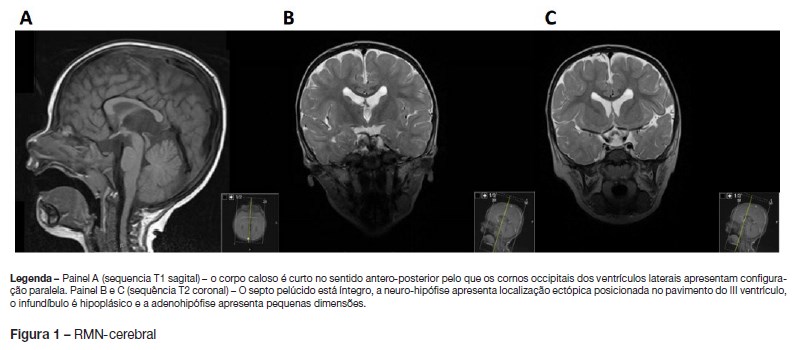
Foi verificada normalização posterior do valor das plaquetas. Realizou RMN-cerebral com pedido de visualização e des- crição da hipófise, que mostrou ectopia da neuro-hipófise, hipoplasia da adeno-hipófise e do infundíbulo assim como corpo caloso curto (Figura 1).
Perante a hipótese de hipoglicemia em contexto de hipopi- tuitarismo colheu doseamentos hormonais, onde se constata- ram défices hipofisários (Quadro 2) de ACTH e GH o que confir- mou o diagnóstico.
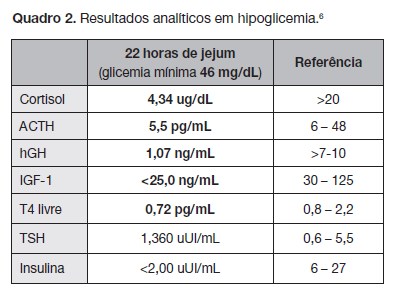
Iniciou terapêutica de substituição com hidrocortisona (10 mg/m2/dia). Após 3 semanas foi confirmado défice de TSH (TSH 1,275 uUI/mL dentro do valor de referência, T4 livre 0,70 pg/mL – inferior ao valor de referência) tendo iniciado levotiroxina (2 µg/ kg/dia) – dado que o défice de ACTH pode mimetizar um falso hipotiroidismo.7
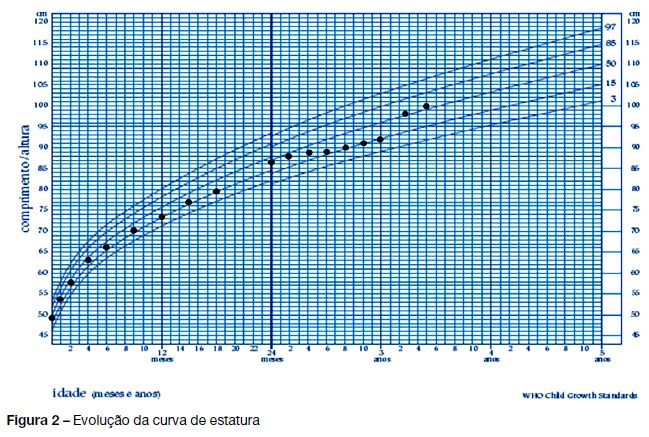
Após 6 meses, foi constatada diminuição da velocidade de crescimento (VC), inferior ao P3 (Figura 2) pelo que foi iniciado tratamento comparticipado com somatotropina (0,025 mg/kg/ dia), após aprovação pela Comissão Nacional para a Normalização da Hormona do Crescimento.
Actualmente, com 4 anos de idade, mantém-se em segui- mento trimestral em consulta de Endocrinologia Pediátrica, apresentando crescimento regular com recuperação de VC e desenvolvimento psicomotor adequado. Mantem terapêutica com hidrocortisona, levotiroxina e somatotropina.
DISCUSSÃO
O diagnóstico de hipopituitarismo exige elevado grau de suspeição.
No período neonatal a ocorrência de hipoglicemia persistente e icterícia colestática deveriam ter levantado esta hipótese.
Nesta criança a descompensação surgiu no decurso de uma primeira intercorrência infecciosa, o que se deve a insuficiência adrenal.
A ocorrência de défices hormonais habitualmente é progressiva, obrigando à avaliação clínica e/ou laboratorial regular de todos os eixos hipofisários.1,4 Neste caso será de esperar a necessidade de terapêutica de substituição com hormonas sexuais na adolescência, caso se venha a confirmar hipogonadismo central.
O tratamento do hipopituitarismo exige reposição hormonal durante toda a vida.1-3 A adesão terapêutica deve ser consistente e minuciosa, uma vez que o prognóstico será favorável se o tratamento for adequado e precoce.1-3
A neuroimagem é fulcral para a detecção de anomalias estruturais.8
São várias as moléculas de sinalização e factores de trans- crição implicados na organogénese e diferenciação celular hipofisária.9 O fenótipo é determinado pela mutação genética, que pode causar múltiplos défices hormonais (exemplo: PIT1, POUF1, PROP1, LHX3, LHX4, HESX1) ou, menos comumente, um défice isolado.1-3,9
Perante uma hipoglicemia grave, deve ser sempre investiga- da a etiologia. O estudo inicial deve incluir a avaliação da função hipofisária e deve ser realizado de preferência nas primeiras colheitas (tal como foi realizado neste caso) após a hipoglicemia para não protelar o diagnóstico.
EM DESTAQUE
O diagnóstico de hipopituitarismo exige elevado grau de suspeição. No período neonatal a ocorrência de hipoglicemia persistente e icterícia colestática deveria ter levantado esta hipótese de diagnóstico. A ocorrência dos défices hormonais habitualmente é progressiva, obrigando à avaliação clínica e laboratorial regular de todos os eixos hormonais. A neuroimagem é fulcral para a deteção de anomalias estruturais associadas. Nesta criança a descompensação surgiu no decurso de uma infeção, como é espetável na insuficiência adrenal. Perante uma hipoglicemia, deve ser sempre investigada a etiologia, o que deve incluir estudo de função hipofisária.
HIGHLIGHTS
The diagnosis of hypopituitarism requires high degree of suspicion. In the neonatal period the occurrence of persistent hypoglycemia and cholestatic jaundice should have raised this diagnosis. The occurrence of hormonal deficits is progressive, which demands regular clinical and laboratory evaluation of all hormonal axes. Neuroimaging is important to detect structural anomalies. In this child decompensation arose during an infection, which is normal in adrenal insufficiency. Investigation of hypoglycemia´s etiology should always include study of pituitary function.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Brook C, Clayton P, Brown R. Brook´s Clinical Pediatric Endocrinology. Quinta Edição. Oxford. Blackwell Publishing. 2005. [ Links ]
2. Ascoli P, Cavagnini F. Hypopituitarism. Pituitary. 2006; 9:335-42. [ Links ]
3. De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, et al. Hypopituitarism. Endotext – Publicação Online. South Dartmouth (MA): MDText.com, Inc.; 2000-15. [ Links ]
4. Cavarzere P, Biban P, Gaudino R, Perlini S, Sartore L, Chini L, et al. Diagnostic pitfalls in the assessment of congenital hypopituitarism. Journal of Endocrinological Investigation. 2014; 37:1201-9. [ Links ]
5. Toogood A, Stewart P. Hypopituitarism: clinical features, diagnosis, and management. Endocrinology & Metabolism Clinics of North America. 2008; 37:235-61. [ Links ]
6. Esoterix Endocrinology Expected Values and S.I. Unit Conversion Tables. Laboratory Corporation of America (2011). [ Links ]
7. Melmed S, Polonsky KS, Larsen PR, Kronenberg HM. Williams Textbook of Endocrinology. 12ª Ed. Saunders. 2012. [ Links ]
8. Tsai S, Laffan E, Lawrence S. A retrospective review of pituitary MRI findings in children on growth hormone therapy. Pediatric Radiology. 2012; 42:799-804. [ Links ]
9. Larson A, Nokoff NJ, Meeks NJ. Genetic causes of pituitary hormone deficiencies. Discov Med. 2015; 19:175-83. [ Links ]
Endereço para correspondência
Maria Miguel Gomes
Serviço de Pediatria
Hospital de Braga
Sete Fontes
4710-243 São Victor, Braga
Email: mariamgomes@hotmail.com
Recebido a 05.01.2015 | Aceite a 29.12.2015


{kind=link}
{kind=link}