










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.26 no.2 Porto jun. 2017
CASE REPORTS | CASOS CLÍNICOS
Instabilidade cromossómica e imunodeficiência - associação essencial no diagnóstico de Síndrome de Nijmegen
Chromosomal instability and immunodeficiency – essential for the diagnosis of Nijmegen Syndrome
Joana CorreiaI; Liliana PinhoI; Isabel Couto GuerraI; Emília CostaI; Esmeralda CletoI
I Pediatric Hematology Unit, Department of Pediatrics, Centro Materno Infantil do Norte, Centro Hospitalar do Porto. 4050-371 Massarelos, Portugal. jbcorreia@sapo.pt; liliana.pinho@gmail.com; isabelcoutoguerra@sapo.pt; emvcosta@hotmail.com; esmeraldacleto@gmail.com
RESUMO
Introdução: O Síndrome de Quebras de Nijmegen (SQN), é uma doença autossómica recessiva rara, pertencente ao grupo dos síndromes de instabilidade cromossómica, sendo mais pre- valente na europa central e do leste.
Caso clínico: Descreve-se o caso de um rapaz de 14 meses, filho de pais ucranianos, que nasceu pequeno para a idade gestacional, com microcefalia e dismorfias faciais, que se tornaram mais evidentes com o crescimento.
O estudo complementar revelou instabilidade cromossómica espontânea e induzida, hemoglobina fetal e α-fetoproteína normais e imunodeficiência celular. Estas características sugeriram o diagnóstico de SQN, confirmado pela identificação da mutação fundadora (657del5) em homozigotia, comum na população eslava.
Discussão: O SQN deve ser considerado na presença de microcefalia, características faciais típicas, atraso de crescimento, instabilidade cromossómica e imunodeficiência. O prognóstico é desfavorável pela ocorrência de infeções de repetição e elevada incidência de neoplasias. O seguimento multidisciplinar e a evicção da exposição a radiação ionizante são fundamentais.
Palavras-chave: Síndrome de Quebras de Nijmegen; instabilidade cromossómica; imunodeficiência
ABSTRACT
Introduction: Nijmegen breakage syndrome (NBS) is a rare autosomal recessive disorder that belongs to the group of chromosomal instability syndromes, more prevalent in Central and Eastern Europe.
Clinical case: We describe the case of a 14-month-old boy, born to Ukrainian parents, that presented at birth with microcephaly, small to gestational age and dysmorphic facial characteristics, which became more obvious with age. The complementary study revealed spontaneous and induced chromosomal instability, with normal fetal haemoglobin and α-fetoprotein and cellular immunodeficiency. These features suggested NBS, confirmed by the identification of the founder mutation (657del5) in homozygous state, common in patients of Slavic origin.
Discussion: NBS should be considered in the presence of microcephaly, typical facial features, growth retardation, chromosomal instability and immunodeficiency. The prognostic is poor due to the occurrence of frequent infections and the high incidence of cancer. Multidisciplinary follow-up and avoidance of radiation exposition are crucial.
Keywords: Nijmegen Breakage Syndrome; chromosomal instability; immunodeficiency
INTRODUÇÃO
Os síndromes de instabilidade cromossómica (SIC) são um conjunto de doenças raras, habitualmente de transmissão autossómica recessiva, que se caracterizam por um aumento espontâneo ou induzido da frequência de quebras e/ou rearranjos cromossómicos, resultando numa maior susceptibilidade à ocorrência de neoplasias. Os exemplos clássicos são a Anemia de Fanconi (AF), a Ataxia-Telangiectasia (AT) e o Síndrome de Bloom (SB). No entanto, nas últimas décadas têm sido incluídas novas doenças.1
Descrito inicialmente em 1981, o Síndrome de Quebras de Nijmegen (SQN) resulta de mutações no gene NBN, localizado no cromossoma 8 (8q21), que codifica a nibrina - proteína indispensável ao processo de reparação do ADN.2 Manifesta-se por instabilidade cromossómica associada a imunodeficiência. Presente a nível mundial, a sua incidência e prevalência são desconhecidas, no entanto, a maioria dos doentes provém da europa central e do leste, devido à presença de uma mutação fundadora.3
CASO CLÍNICO
Primeiro filho de um casal ucraniano, não consanguíneo, com antecedentes familiares irrelevantes. A gravidez foi complicada por restrição do crescimento fetal detetada às 34 semanas de gestação, nascendo às 39 semanas por parto eutócico. Na observação inicial salientava-se um peso de 2500g (percentil (P) 2), comprimento 45 cm (P1), perímetro cefálico de 31cm (P1) e uma fácies peculiar.
Nos primeiros meses de vida verificou-se um agravamento progressivo da microcefalia, com desenvolvimento psicomotor normal e recuperação estaturoponderal. Sem outros antecedentes patológicos relevantes. Durante a investigação da microcefalia foi realizado um cariótipo de sangue periférico, detetando-se a presença de quebras cromossómicas espontâneas.
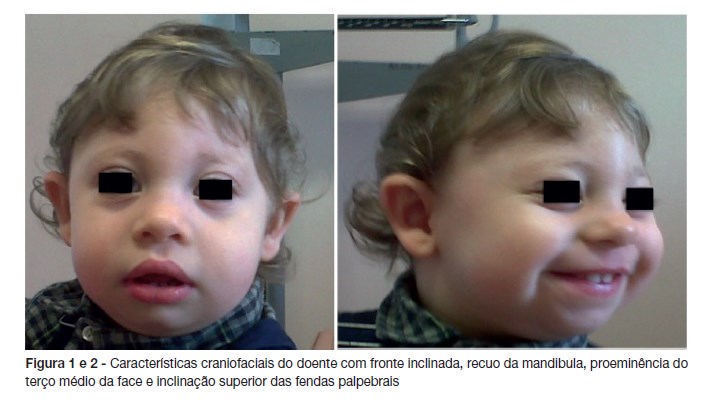
Pela suspeita de AF foi referenciado aos 14 meses à consulta de Hematologia Pediátrica. Adicionalmente, de salientar que apresentava o Programa Nacional de Vacinação atualizado, sem vacinas extra-programa, e sem registo de reações adversas. No exame objetivo apresentava uma microcefalia grave (40 cm; <P1, SDS -4,86), proeminência do terço médio da face, hipertelorismo e inclinação superior das fendas palpebrais (Figura 1 e 2). O peso e comprimento encontravam-se nos percentis 10-25 e 25, respetivamente. Apresentava uma mancha café-au-lait na região lombar direita, sem outras alterações cutâneas, e o exame neurológico era normal.
O estudo complementar efetuado confirmou a presença de quebras cromossómicas espontâneas e induzidas por diepoxibutano (DEB). No hemograma apresentava uma linfopenia ligeira (1280/μL), com as restantes linhas celulares e esfregaço de sangue periférico sem alterações. Os doseamentos de hemo- globina fetal e α-fetoproteina foram normais (1,4% e 12,7μg/L respetivamente). A avaliação ultrassonográfica abdominal e renal não identificou alterações.
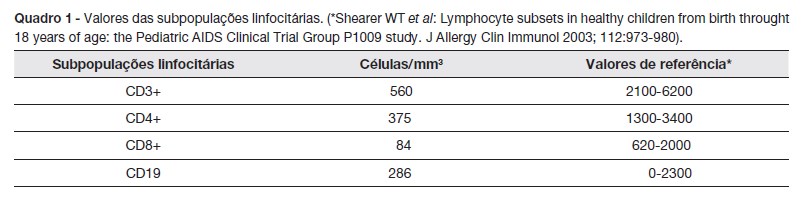
A imunofenotipagem dos linfócitos de sangue periférico (Quadro 1) revelou uma linfopenia B e T (CD3 560/mm3; CD4 375/mm3; CD8 84/mm3, CD4/CD8 4,4; CD19 286/mm3) com aumento das células CD4+ memória, em detrimento das células CD4+ naive (CD45RA 65% e CD45RO 34%). O doseamento das imunoglobulinas séricas (IgA, IgG e IgM) e subclasses de IgG foi normal.
A conjugação dos dados analíticos (instabilidade cromossómica e imunodeficiência celular), dismorfológicos (microcefalia grave associada a características faciais típicas) e epidemiológicos (origem étnica dos progenitores) permitiu equacionar o diagnóstico de SQN, confirmado pelo estudo genético – identificação da mutação fundadora (c.657_661delACAAA) em homozigotia.
Aos quatro anos de idade, o doente mantem uma evolução de peso e estatura nos P25 e P10-25, apresentando uma perturbação do desenvolvimento da linguagem; sem intercorrências infeciosas graves ou recorrentes. Analiticamente, evidencia uma linfopenia grave (735/μL linfócitos, CD4 256/mm3), sem imunodeficiência humoral.
Desde o diagnóstico, mantém seguimento multidisciplinar pelas Unidades de Hematologia, Desenvolvimento e Imunodeficiências. Foi realizada vacinação antipneumocócica conjugada 13-valente, antimeningocócica tipo B e antigripal, e dada a linfopenia T grave foi instituída profilaxia com cotrimoxazol.
DISCUSSÃO
O reconhecimento do SQN reside na identificação da associação de dismorfias características, imunodeficiência e instabilidade cromossómica, sobretudo perante indivíduos oriundos de áreas geográficas com prevalência aumentada.
A sua principal característica fenotípica é uma microcefalia grave, progressiva, que está presente ao nascimento em cerca de 75% dos doentes, desenvolvendo-se nos primeiros meses de vida nos restantes casos4. Além disso, os doentes apresentam dismorfias faciais típicas que se tornam mais evidentes com o crescimento, sendo facilmente reconhecidas por volta dos três anos de idade. Tal como no caso descrito, a fronte está inclinada e há um recuo da mandíbula, com proeminência do terço médio da face (face de pássaro). Podem ocorrer outras características faciais mais subtis, como uma inclinação superior das fendas palpebrais ou pirâmide nasal longa e pontiaguda ou com narinas antevertidas3.
Em cerca de 50% dos doentes estão presentes, outras malformações congénitas, incluindo clinodactilia e/ou sindactilia, fenda do palato, atrésia das coanas, displasia da anca, atrésia anal, hipospadias, criptorquidia, hipoplasia/agenesia renal ou rim em ferradura ou duplo. As manifestações cutâneas incluem manchas café-au-lait, vitiligo e múltiplos nevos pigmentados.3,4
Em termos de evolução estatural, os doentes podem ser pequenos para a idade gestacional ao nascimento e o atraso de crescimento é comum nos primeiros dois/três anos de vida. Posteriormente, a velocidade de crescimento normaliza até à adolescência, altura em que o surto de crescimento pubertário está diminuído no sexo masculino e ausente no feminino.5 Os indivíduos do sexo feminino têm insuficiência gonadal primária.6 Para além da microcefalia, os doentes apresentam uma diminuição do tamanho dos lobos frontais e estreitamento dos cornos frontais dos ventrículos laterais. Esta alteração parece resultar de um desenvolvimento cerebral subótimo (em particular dos lobos frontais), associado a um encerramento prematuro das fontanelas e suturas. A agenesia da porção posterior do corpo caloso associada a colpocefalia e dilatação dos cornos temporais é outra anomalia frequente do sistema nervoso central.7,8 Apesar destas alterações estruturais, não existem habitualmente outras manifestações neurológicas e os marcos do desenvolvimento são atingidos nas idades normais durante o primeiro ano de vida4. Contudo, posteriormente, torna-se característico um atraso do desenvolvimento da linguagem com necessidade de intervenção terapêutica e durante a idade escolar a maioria das crianças apresenta um défice intelectual ligeiro-moderado.3 A presença de imunodeficiência típica deste síndrome, traduz-se clinicamente pela ocorrência de infeções sinopulmonares recorrentes na maioria dos doentes, nomeadamente otite média aguda, mastoidite, sinusite e pneumonia, estando descrita a evolução para doença pulmonar crónica com bronquiectasias. As infeções urinárias e gastrointestinais são também comuns.3
Os distúrbios imunológicos variam de doente para doente e num mesmo indivíduo ao longo do tempo, podendo englobar a imunidade celular e/ou humoral.3 A imunodeficiência humoral caracteriza-se por défice de um ou mais isótipos de imunoglobulina ou subclasses de IgG (principalmente IgG2 e/ou IgG4), mesmo que a concentração total de IgG seja normal. A agamaglobulinemia está presente em cerca de 20% dos doentes.9
Dentro dos defeitos da imunidade celular salienta-se a diminuição do número absoluto de linfócitos CD3+ e CD4+, existindo habitualmente, ao contrário do caso descrito, uma redução da relação CD4/CD8 (<1). O número de linfócitos T CD8+, habitualmente está inalterado, mas pode estar aumentado ou muito diminuído. Ocorre também um aumento das células T memória (CD45RO+) com consequente redução das células naive (CD45RA+). O número absoluto de células B (CD19+) está habitualmente reduzido, mas poderá ser normal ou até elevado em alguns doentes.9,10
Finalmente salienta-se a presença de instabilidade cromossómica, resultante da disfunção do sistema de reparação de ADN e traduzida pelo aumento da susceptibilidade ao cancro.4 A incapacidade de reconhecer e reparar danos a nível do ADN pode resultar em morte celular prematura ou transformação maligna. Os doentes apresentam múltiplos rearranjos, envolvendo principalmente translocações e inversões entre os cromossomas 7 e 14.13
O estudo citogenético do sangue periférico geralmente permite detetar instabilidade cromossómica espontânea, embora a má resposta aos mitogénios, com consequente baixo índice mitótico, característica do SQN, possa dificultar a avaliação.3 Os doentes apresentam hipersensibilidade cromossómica à radiação ionizante e agentes radiomiméticos (p. ex. bleomicina), com aumento da frequência de rearranjos cromossómicos.11
A reação a agentes alquilantes (DEB ou mitomicina C) é variável, mas pode haver hipersensibilidade moderada, levando a uma evocação diagnóstica inicial, tal como no presente caso, ou até mesmo assunção diagnóstica errónea de AF.12,13 A microcefalia, as alterações cutâneas, o atraso do crescimento e a susceptibilidade aumentada a neoplasias são manifestações comuns às duas entidades. Porém, a imunodeficiência associada à ausência de marcadores de eritropoiese de stress (macrocitose e aumento da HbF) e de outras citopenias, para além da linfopenia, sugerem outro diagnóstico, que não a AF.13
O SQN deve ser ainda diferenciado de outros síndromes de instabilidade cromossómica. Relativamente à AT, ambos apresentam imunodeficiência combinada, sensibilidade à radiação ionizante e rearranjos cromossómicos típicos entre os cromossomas 7 e 14. No entanto no SQN estão ausentes as manifestações neurocutâneas (ataxia, apraxia e telangiectasias) características da AT e a α-fetoproteina está normal.12 O síndrome Ligase IV apresenta um fenótipo semelhante ao SQN, mas resulta de mutações num gene distinto que codifica a ADN ligase IV, localizado no cromossoma 13q33. Os doentes apresentam habitualmente microcefalia, dismorfias faciais, atraso de crescimento e desenvolvimento, mas tipicamente evidenciam pancitopenia.14
O diagnóstico definitivo de SQN estabelece-se pelo estudo molecular, com a demonstração da existência de uma mutação no gene NBN. Na população de origem eslava, na qual se inclui o doente descrito, a sequenciação do exão 6 é o método de eleição dada a elevada prevalência da mutação fundadora 657_del5 (1/177 habitantes).15
Excluindo as mutações de novo, os pais de uma criança afetada são portadores de mutações, com um risco de 25% de transmissão da doença em cada gravidez, devendo ser disponibilizado aconselhamento genético. Além disso, apesar de assintomáticos, os indivíduos heterozigóticos possuem um risco acrescido de neoplasia (em particular, os portadores da mutação 657_del5), pelo que deverão manter vigilância clínica regular.3
Não existe tratamento específico para o SQN, sendo o diagnóstico precoce e o seguimento multidisciplinar a longo-prazo fundamentais. A avaliação imunológica e, quando indicadas, a terapêutica terapêutica substitutiva com Ig e/ou profilaxia antimicrobiana, são importantes na prevenção de infeções graves recorrentes e suas complicações. Por outro lado, a evicção da exposição desnecessária à radiação ionizante para fins diagnósticos ou terapêuticos é igualmente mandatória.3,15 As doentes do sexo feminino deverão ser orientadas por Endocrinologia em idade pubertária.3
O prognóstico deste síndrome é mau, sendo as doenças malignas a principal causa de morte. Cerca de 40% dos doentes desenvolvem uma neoplasia antes dos 20 anos. As mais comuns são os linfomas de células T e B; no entanto, estão também descritos casos de gliomas, rabdomiossarcomas e meduloblastomas.3
Em conclusão, perante um doente com evidência de um síndrome de instabilidade cromossómica, mesmo perante um fenótipo compatível, não se deve assumir o diagnóstico mais prevalente (AF), sobretudo quando estão presentes dados não típicos (neste caso, linfopenia isolada perante a ausência de marcadores de eritropoiese de stress). Nesses casos, a conjugação de todos os elementos clínicos disponíveis é crucial.
AGRADECIMENTOS
À Prof.ª Doutora Beatriz Porto, diretora do Laboratório de Citogenética do Instituto de Ciências Biomédicas Abel Salazar, pela sua colaboração na avaliação de instabilidade cromossómica.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Taylor AMR. Chromosome instability syndromes. Best Practice Research Clinical Haematology. 2001; 14:631-44 [ Links ]
2. Weemaes CM, Hustinx TW, Scheres JM, van Munster PJ, Bakkeren JA, Taalman RD. A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr Scand. 1981; 70:557–64. [ Links ]
3. Chrzanowska KH, gregorek H, Dembowska-Baginska B, Kalina MA, Digweed M. Nijmegen Breakage Syndrome. Orphanet Journal of Rare Diseases. 2012; 7:13. [ Links ]
4. The International Nijmegen Breakage Syndrome Study Group. Nijmegen Breakage Syndrome. Arch Dis Child. 2000; 82:400-6. [ Links ]
5. Chrzanowska K, Kalina M, Rysiewski H, Gajdulewicz M, Szarras-Czapnik M, Gajtko-Metera M, et al. Growth pattern in patients with Nijmegen breakage syndrome: evidence from a longitudinal study [abstract]. Horm Res Paediatr. 2010; 74(Suppl 3):s38. [ Links ]
6. Chrzanowska KH, Szarras-Czapnik M, Gajdulewicz M, Kalina MA, Gajtko-Metera M, Walewska-Wolf M, et al. High prevalence of primary ovarian insufficiency in girls and young women with Nijmegen breakage syndrome: evidence from a longitudinal study. J Clin Endocrinol Metab. 2010; 95:3133-40. [ Links ]
7. Bekiesińska-Figatowska M, Chrzanowska KH, Sikorska J, Walecki J, Krajewska-Walasek M, Jóźwiak S. Cranial MRI in the Nijmegen breakage syndrome. Neuroradiology. 2000; 42:43–7. [ Links ]
8. Bekiesińska-Figatowska M, Chrzanowska KH, Jurkiewicz E, Wakulińska A, Rysiewski H, Gładkowska-Dura M, et al. Magnetic resonance imaging of brain anormalities in patients with the Nijmegen breakage syndrome. Acta Neurobiol Exp. 2004; 64:503-9. [ Links ]
9. Gregorek H, Chrzanowska KH, Michalkiewicz J, Syczewska M, Madalinski K. Heterogeneity of humoral immune abnormalities in children with Nijmegen breakage syndrome: an 8-year follow-up study in a single centre. Clin Exp Immunol. 2002; 130:319–24. [ Links ]
10. Gregorek H, Chrzanowska KH, Dzierżanowska-Fangrat K, Wakulińska A, Pietrucha B, Zapaśnik A, et al. Nijmegen breakage syndrome: long-term monitoring of viral and immunological biomarkers in peripheral blood before development of malignancy. Clin Immunol. 2010;135: 440–7. [ Links ]
11. Pérez-Vera P, González-del Angel A, Molina B, Gómez L, Frías S, Gatti RA, et al. Chromosome instability with bleomycin and X-ray hypersensitivity in a boy with Nijmegen breakage syndrome. Am J Med Genet. 1997; 70:24–7. [ Links ]
12. Chrzanowska KH, Stumm M, Bekiesińska-Figatowska M, Varon R, Białecka M, Gregorek H, et al. Atypical clinical picture of the Nijmegen breakage syndrome associated with developmental abnormalities of the brain. J Med Genet. 2001; 38:E3. [ Links ]
13. Gennery AR, Slatter MA, Bhattacharya A, Barge D, Haigh S, ODriscoll M, et al. The clinical and biological overlap between Nijmegen Breakage Syndrome and Fanconi anemia. Clin Immunol. 2004; 113:214–9. [ Links ]
14. Ben-Omran TI, Cerosaletti K, Concannon P, Weitzman S, Nezarati MM. A patient with mutations in DNA Ligase IV: clinical features and overlap with Nijmegen breakage syndrome. Am J Med Genet A. 2005; 137A:283–7. [ Links ]
15. Varon R, Seemanova E, Chrzanowska K, Hnateyko O, Piekutowska-Abramczuk D, Krajewska-Walasek M, et al. Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations. Eur J Hum Genet. 2000; 8:900–2. [ Links ]
CORRESPONDENCE TO
Joana Correia
Pediatric Hematology Unit
Department of Pediatrics
Centro Materno Infantil do Norte
Centro Hospitalar do Porto
Largo da Maternidade Júlio Dinis s/n 4050-371 Massarelos
Email: jbcorreia@sapo.pt
Received for publication: 22.06.2016 Accepted in revised form: 26.09.2016


{kind=link}
{kind=link}