










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.28 no.3 Porto set. 2019
https://doi.org/10.25753/BirthGrowthMJ.v28.i3.15338
CASE REPORTS | CASOS CLÍNICOS
Severe hypokalemia in a child with mild gastroenteritis
Hipocalémia grave numa criança com gastroenterite ligeira
Catarina FariaI, Carina FerreiraII, Alzira SarmentoIII, Sara GonçalvesIII, Paula RochaIII, Carlos DuarteIV, Conceição MotaV
I. Department of Pediatrics, Hospital de Braga. 4710-243 Braga, Portugal. catmagalhaesfaria@gmail.com
II. Department of Pediatrics, Hospital de São Teotónio. 3504-509 Viseu, Portugal. casferreirast@gmail.com
III. Pediatric Intensive Care Unit, Centro Materno-Infantil do Norte, Centro Hospitalar Universitário do Porto. 4099-001 Porto, Portugal. sarmento.alzira@gmail.com; sara_g_goncalves@hotmail.com; paulammrocha@gmail.com
IV. Department of Pediatrics, Hospital da Luz Arrábida. 4400-346 Vila Nova de Gaia, Portugal. dcarlosduart@sapo.pt
V. Department of Pediatric Nephrology, Centro Materno-Infantil do Norte, Centro Hospitalar Universitário do Porto. 4099-001 Porto, Portugal. conceicaocmota@gmail.com
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
Background: Hypokalemia (serum potassium below 3.5 mmol/L) may be caused by several mechanisms. Severe hypokalemia must be immediately managed, as it can have important cardiac repercussions.
Clinical case: A previously healthy eight-year-old female, with normal growth and normal-to-low blood pressure, was observed due to persistent abdominal pain, anorexia, and fever two days after overcoming a mild gastroenteritis episode. Serum biochemistry revealed severe hypokalemia (1.8 mmol/L), hypomagnesemia, and metabolic alkalosis. The patient was admitted to the Pediatric Intensive Care Unit for correction of electrolyte imbalance, cardiac monitoring, and investigation. Complementary studies included a spot urine ionogram that revealed inappropriate potassium wasting. Ionic correction was achieved by both intravenous and oral supplementation plus spironolactone. Genetic testing was positive for Gitelman syndrome.
Discussion/Conclusion: Suspicion of complex causes should be raised and a comprehensive approach undertaken upon a discrepancy between clinical history and hypokalemia severity.
Keywords: electrolyte imbalance; hypokalemia; Gitelman syndrome; tubulopathy
RESUMO
Introdução: A hipocalémia (potássio sérico inferior a 3.5 mmol/L) pode ser causada por vários mecanismos. A hipocalémia grave é uma emergência médica pelas importantes repercussões cardíacas que pode ter.
Caso clínico: Uma rapariga de oito anos, previamente saudável, com crescimento normal e tensão arterial tendencialmente baixa, foi observada por dor abdominal persistente, anorexia e febre dois dias após recuperar de uma gastroenterite aguda ligeira. A avaliação bioquímica revelou hipocalémia grave (1.8 mmol/L), hipomagnesémia e alcalose metabólica. A rapariga foi admitida na Unidade de Cuidados Intensivos Pediátricos para correção dos distúrbios eletrolíticos, monitorização cardíaca e investigação. O ionograma urinário (urina pontual) revelou excreção inapropriada de potássio. A correção iónica foi alcançada através de suplementação intravenosa e oral associadas a espironolactona. A pesquisa de mutações para Síndrome de Gitelman foi positiva.
Discussão/Conclusão: Perante uma discrepância entre o quadro clínico e a gravidade da hipocalémia, deverão ser suspeitadas e pesquisadas 155 causas complexas deste distúrbio eletrolítico.
Palavras-chave: distúrbios eletrolíticos; hipocalémia; síndrome de Gitelman; tubolopatia
Introduction
Hypokalemia (serum potassium below 3.5 mmol/L) may be caused by excessive (gastrointestinal, renal, or cutaneous) potassium loss, increased intracellular movement, and/or decreased intake (Table 1).1,2 Two or more underlying mechanisms may be present, especially in cases of severe hypokalemia, which should be immediately managed as the condition may have important cardiac consequences, including ventricular fibrillation and subsequent cardiac arrest.2
Common hypokalemia causes, such has diarrhea, vomiting, or diuretic therapy, are usually promptly identified through clinical history. Salt-losing tubulopathies represent a rare cause of hypokalemia and may require special consideration and analysis.2,3
The authors present the case of an eight-year-old girl with severe hypokalemia detected during the course of a mild gastroenteritis, leading to the investigation of other causes of potassium loss.
Case report
An eight-year-old female presented with persistent abdominal pain and anorexia two days after overcoming a mild gastroenteritis episode. The episode lasted for five days and featured small-volume, semi-liquid stools (four episodes in the first two days), sporadic vomiting (two episodes), abdominal cramps, and anorexia. Except for a trend towards low blood pressure, maternal report of usually high urinary output, and a preference for salty food since young age, the child was otherwise previously healthy and had normal growth and psychomotor development. Family history was also unremarkable, with no parental consanguinity.
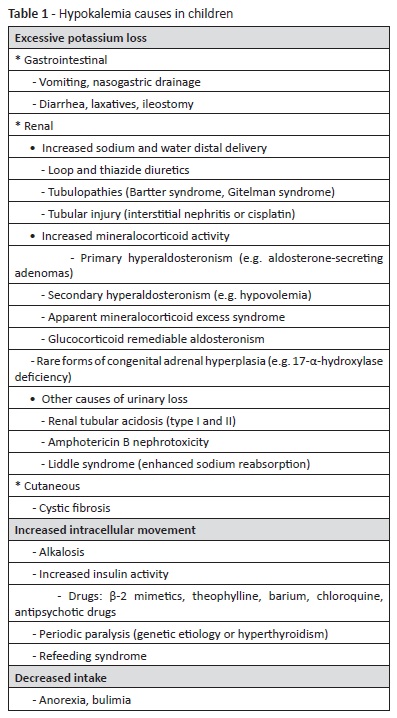
On admission, physical examination revealed low fever (38.6ºC), pale skin and dry mucous membranes, with no other abnormalities. Serum biochemistry found severe hypokalemia (1.8 mmol/L), hyponatremia (130 mmol/L) with hypochloremia (84 mmol/L), hypophosphatemia (0.66 mmol/L), discreet hypomagnesemia (0.58 mmol/L), and metabolic alkalosis (pH 7.53, pCO2 35.6 mmHg, bicarbonate 29.7 mmol/L, base excess 6.4 mmol/L). Blood urea nitrogen and creatinine were within the normal range, and complete blood count and remaining biochemistry were unremarkable. Electrocardiogram (ECG) showed long QT interval (0.53 seconds), ST depression, shallow T waves and prominent U waves, typical findings when hypokalemia is present - Figure 1, Panel A. Abdominal ultrasound was normal. The girl was admitted to the Pediatric Intensive Care Unit for correction of electrolyte imbalance, cardiac monitoring, and investigation.
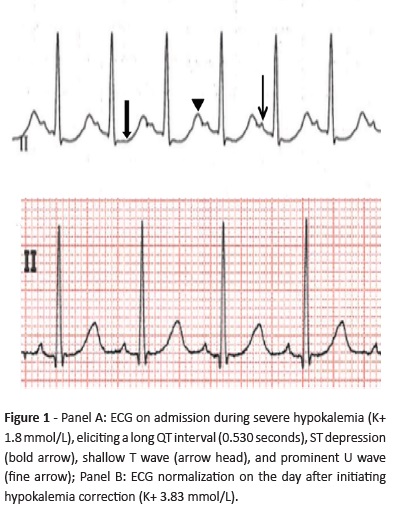
Complementary studies during hospital stay included a spot urine ionogram, which revealed an inappropriate renal potassium wasting (potassium-to-creatinine ratio of 49 mmol/g [normal values <18 mmol/g]), normal calcium excretion (calcium-creatinine ratio of 0.04 mg/mg [normal values 0.01−0.25 mg/mg]), normal phosphorus excretion (phosphorus-creatinine ratio of 0.35 mg/mg [normal values 0.32−0.97 mg/mg]), and normal chloride excretion (fractional chloride excretion of 0.48% [normal values <0.5%]). Estimated glomerular filtration rate was normal for age (>90 mL/ min/1.73m2). Urine density and osmolality were within the normal range (1015 and 632 mOsm/Kg, respectively). Renal and adrenal gland ultrasound revealed no abnormalities. Aldosterone was within the normal range, but hyperreninemia was present (280.9 pg/mL, normal 1.9−41.2 pg/mL). Although the girl was conscious and capable of tolerating oral medication, potassium correction was initially performed intravenously (at a maximum 0.5 mmol/Kg/h rate), due to the need of a rapid correction to prevent cardiac alterations. Oral supplementation was subsequently initiated, together with a potassium, magnesium, and phosphate-enriched diet. ECG normalized by day two with hypokalemia correction (3.83 mmol/L) - Figure 1, Panel B. As normal potassium values were difficult to maintain with oral supplementation only, spironolactone (1 mg/ Kg/day) was initiated in day four. The patient remained with normal blood pressure and adequate urine output. At discharge (day 14), serum potassium level was 3.47 mmol/L and remaining electrolytes were within the normal range, including magnesium (0.69 mmol/L). The girl currently attends the Nephrology consultation for follow-up and is medicated with potassium chloride and magnesium sulfate oral supplements and spironolactone. Genetic testing was positive for Gitelman syndrome (heterozygosity for SLC12A3 mutation).
In the present clinical case, while initial abdominal complaints, nausea, and vomiting were mild gastroenteritis symptoms, the following persistent abdominal pain and anorexia were probably a consequence of hypokalemia and further exacerbated it. Hypokalemia led to diagnosis of an underlying tubular disorder with potentially serious consequences if left untreated, including cardiac arrest.
Severe hypokalemia must be quickly managed and may require correction through central venous catheter, since peripheral infusions with potassium concentrations above 20 mmol/L are painful. Potassium concentration in infusion must not exceed 40 mmol/L and infusion rate should be below 1 mmol/Kg/hour (maximum 20 mmol/h).2 Potassium chloride is the most common preparation used for potassium supplementation.1 Continuous ECG monitoring is mandatory during correction.4
Gitelman syndrome is one of the most common inherited tubulopathies, transmitted in an autosomal-recessive pattern, with an estimated prevalence of 1 to 10 per 40000.5 It is caused by inactivating mutations in SLC12A3 gene and more than 350 mutations have been identified.6 This gene encodes the thiazide-sensitive sodium chloride cotransporter, expressed in the kidney distal convoluted tubules.5 Consequently, there is an increased water and sodium distal delivery, which are reabsorbed in exchange of potassium and hydrogen, resulting in an excessive urine secretion.2,3 This leads to the characteristic hypokalemia and metabolic alkalosis found in the disorder, which are additionally exacerbated by secondary hyperaldosteronism due to volume depletion.7 Other biochemical alterations include hypocalciuria and hypomagnesemia, which are very suggestive of the referred diagnosis but may be absent.8 Hypomagnesemia also contributes to hypokalemia development.9,10 Although mechanisms underlying hypomagnesemia are still not fully understood, it has been associated with downregulation of a magnesium channel expressed on the distal convoluted tubules (TRPM6), with subsequent renal magnesium loss. Downregulation of this channel is believed to be a consequence of poor activity of thiazide-sensitive sodium chloride cotransporter and secondary hyperaldosteronism.9
Gitelman syndrome is usually diagnosed in late childhood or adolescence and has a significantly variable phenotype, even in patients with identical mutations.3,6,11 The disorder may remain undiagnosed until identified in blood testing for unrelated causes or disclosed by conditions that exacerbate the underlying electrolyte imbalance, such as diarrhea or vomiting.6,12,13 Until then, it may be asymptomatic or present with mild and unspecific symptoms, such as muscular weakness, cramps, paresthesia, fatigue, salt craving, polydipsia and polyuria, abdominal distension and pain, constipation, and vomiting.5-7 Some characteristic ECG alterations found in hypokalemia include prolongation of QT interval, ST depression, shallow T waves, and presence of U waves.2,13 Several cardiac arrhythmias may arise depending on hypokalemia severity, from premature atrial and ventricular beats to ventricular fibrillation.2,13 Although the renin-angiotensin-aldosterone system is activated, blood pressure is usually normal to low, particularly if hypokalemia and hypomagnesemia are severe.14 Growth delay, short stature and late puberty may also occur.6 Hypomagnesemia may lead to chondrocalcinosis, a typical Gitelman syndrome complication in adult age.5,6
Proposed biochemical criteria for Gitelman syndrome include chronic hypokalemia (<3.5 mmol/L) with inappropriate potassium wasting (spot potassium-creatinine ratio >18 mmol/g), metabolic alkalosis, hypomagnesemia (<0.7 mmol/L) with inappropriate renal magnesium wasting (>4% fractional magnesium excretion − not measured in this patient), hypocalciuria (normal spot urine calcium-creatinine ratio varies with age, ranging from 0.01 to 0.25 mg/mg in the 7−17-year age group - this patient was within the normal range: 0.04 mg/mg), high plasma renin activity or levels, >0.5% fractional chloride excretion (this patient was within the normal range: 0.48%), low-to-normal blood pressure, and normal renal ultrasound.6,9 Presence of hypertension, family history of kidney disease with autosomal dominant transmission, renal ultrasound alterations, prenatal history of polyhydramnios and hyperechogenic kidneys, and presentation before the age of three argue against Gitelman syndrome diagnosis and should prompt investigation of other causes.6 Further laboratory findings that may be present in Gitelman syndrome include hypophosphatemia (occasionally), hyponatremia (rarely), and glucose intolerance/insulin resistance (rarely).9
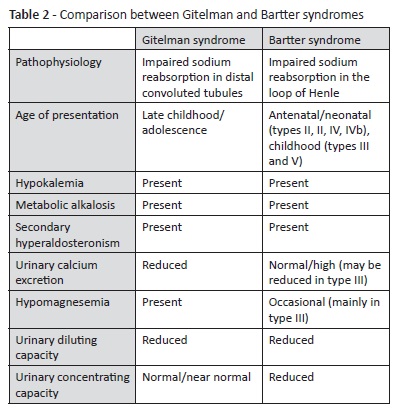
Bartter syndrome, affecting sodium reabsorption in the loop of Henle, is the main inherited pathology to consider in differential diagnosis.3,5 This disorder is often diagnosed during childhood or even in the antenatal or neonatal period, depending on severity.7,12 Some clinical aspects may help to differentiate both syndromes (Table 2). In Bartter syndrome, serum magnesium levels are usually normal, hypercalciuria may be present instead of hypocalciuria, and the ability to concentrate urine is impaired, while in Gitelman syndrome it is preserved.15 However, Bartter syndrome type III, associated with mutations in CLCNKB gene (encoding for chloride channel ClC-Kb located on the distal convoluted tubule), may mimic Gitelman syndrome, presenting with hypomagnesemia and hypercalciuria. Therefore, genetic testing is crucial to establish a definitive diagnosis.5,6
As for non-inherited causes, thiazide diuretic abuse is important to exclude, as these drugs act by blocking the same cotransporter that is defective on Gitelman syndrome, mimicking the disease.7 Other differential diagnoses of Gitelman syndrome include laxative abuse (rare in children) and surreptitious vomiting.3,6
Gitelman syndrome management includes oral potassium supplements, potassium-sparing diuretics and, occasionally, nonsteroidal anti-inflammatory drugs. When hypomagnesemia is present, oral magnesium supplements are also prescribed.3,6,7 Normal serum potassium and magnesium concentrations may be difficult to achieve and maintain with oral supplementation alone. The serum concentration rise following oral supplementation decreases the potassium and magnesium-retaining stimulus, which results from sustained hypokalemia and hypomagnesemia in untreated patients. Consequently, most administered potassium and magnesium is excreted, demanding high supplement doses, which are difficult to tolerate due to side effects.7 A 3.0 mmol/L potassium and 0.6 mmol/L magnesium level are considered satisfactory in Gitelman syndrome.6 Patients should be (at least) annually followed in Nephrology consultation, and growth and puberty carefully assessed.5,6 Genetic counseling should be offered to parents and children.5,6 Patient and caregiver education is key to ensure treatment compliance and promptly identify emergency situations.5,12 Overall, Gitelman syndrome’s long-term prognosis is favorable, although long-term consequences as chronic kidney disease, secondary hypertension, cardiac arrhythmias, and chondrocalcinosis are not totally clarified and should be investigated.5,6
Conclusion
A high index of suspicion of complex causes should be kept upon discrepancy between clinical history and hypokalemia severity, and a comprehensive management approach should be undertaken. Tubulopathies such has Bartter and Gitelman syndrome are often diagnosed in childhood and adolescence, particularly when a common stressful event exacerbates the underlying chronic electrolyte imbalance, unmasking it. Therefore, these syndromes should be considered in hypokalemia differential diagnosis.
REFERENCES
1. Zieg J, Gonsorcikova L, Landau D. Current views on the diagnosis and management of hypokalaemia in children. Acta Paediatr. 2016; 105:762-72. [ Links ]
2. Somers M, Traum A. Hypokalemia in children. [UpToDate website]. August 22, 2016. (Accessed March 20, 2018) Available at: https://www.uptodate.com/contents/hypokalemia-in-children. [ Links ]
3. Koudsi L, Nikolova S, Mishra V. Management of a severe case of Gitelman syndrome with poor response to standard treatment. BMJ Case Rep. 2016 Published online [Feb 17 2016]: DOI: 10.1136/bcr-2015-212375 . [ Links ]
4. Bockenhauer D, Zieg J. Electrolyte disorders. Clin Perinatol. 2014; 41:575-90. [ Links ]
5. Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. 2008; 3:22. [ Links ]
6. Blanchard A, Bockenhauer D, Bolignano D, Calò L, Cosyns E et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017; 91:24-33. [ Links ]
7. Emmet M, Ellison D. Bartter and Gitelman syndromes. [UpToDate website]. February 7, 2018. (Accessed March 20, 2018). Available at: https://www.uptodate.com/contents/ bartter-and-gitelman-syndromes. [ Links ]
8. Colussi G, Bettinelli A, Tedeschi S, De Ferrari M, Syrén M, et al. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol. 2007; 2:454-60. [ Links ]
9. Filippatos T, Rizos C, Tzavella E, Elisaf M. Gitelman syndrome: an analysis of the underlying pathophysiologic mechanisms of acid-base and electrolyte abnormalities. Int Urol Nephrol. 2018; 50:91-6. [ Links ]
10. Huang C, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. 2007;18:2649-52. [ Links ]
11. Lin S, Cheng N, Hsu Y, Halperin M. Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/chloride cotransporter. Am J Kidney Dis. 2004; 43:304-12. [ Links ]
12. Fremont O, Chan J. Understanding Bartter syndrome and Gitelman syndrome. World J Pediatr. 2012; 8:25-30. [ Links ]
13. Cortesi C, Lava SA, Bettinelli A, Tammaro F, Giannini O, Caiata- Zufferey M, et al. Cardiac arrhythmias and rhabdomyolysis in Bartter-Gitelman patients. Pediatr Nephrol. 2010; 25:2005-8. [ Links ]
14. Cruz D, Simon D, Nelson-Williams C, Farhi A, Finberg K, Burleson L, et al. Mutations in the Na-Cl cotransporter reduce blood pressure in humans. Hypertension. 2001; 37:1458-64. [ Links ]
15. Bettinelli A, Bianchetti MG, Girardin E, Caringella A, Cecconi M, Appiani AC, et al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr. 1992; 120:38-43. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Catarina Faria
Department of Pediatrics,
Hospital de Braga
Lugar das Sete Fontes
4710-243 São Victor, Braga
Email: catmagalhaesfaria@gmail.com
Received for publication: 06.11.2018
Accepted in revised form: 14.02.2019

