










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.1 Porto jan. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i1.15336
CASE REPORTS | CASOS CLÍNICOS
Bilateral congenital semicircular canal malformation and hearing loss − case report
Malformação congénita bilateral dos canais semicirculares e perda auditiva - descrição de casos
Joana Raquel CostaI, Miguel CoutinhoI, Teresa SoaresI, Cecília Almeida e SousaI
I. Department of Otolaryngology Head and Neck Surgery, Centro Hospitalar Universitário do Porto. 4099-001 Porto, Portugal. joana_cccosta@hotmail.com; mb.coutinho@gmail.com; teres.soares.costa@gmail.com; director.orl@hgsa.min-saude.pt
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
The main aims of this observational study were to describe a poorly characterized malformation of the inner ear termed bilateral congenital semicircular canal malformation; determine if the degree and pattern of semicircular canal dysmorphology and the presence or absence of a well-formed cochlea predict audiological outcomes, type, and severity of congenital hearing loss; and investigate its relationship with known syndromic forms of hearing loss. Review of eight cases of hearing loss with radiographic evidence of congenital semicircular canal malformation was performed. Information was collected on clinical history, physical examination, computed tomography study and serial audiograms for all patients. Analyzed features included other syndrome-characteristic phenotypic dysmorphologies, audiometric configuration, severity and type of hearing loss, type of audiological rehabilitation, and presence of associated inner ear abnormalities besides those in the vestibular system.
Among the eight cases included in the study, six patients had recognized syndromes/chromosomal abnormalities. Hearing loss was moderate to profound in all cases. All patients had bilateral semicircular canal deformities, with usually identical anatomical pattern on each side. Of the eight cases, six had normal cochlear development; malformations in the tympanic membrane and external auditory canal were only found in one; changes in ossicular chain were found in three patients; vestibules and vestibular aqueduct were normal in most cases; and abnormalities of oval window development and hypoplasia were found in two cases.
The present study shows that a correlation between the severity and type of hearing loss and radiographic abnormalities is difficult to establish. Hearing loss associated with semicircular canal dysplasia is more likely due to anomalous membranous labyrinth development, which is not radiologically detectable by computerized tomography scan.
Keywords: congenital abnormalities; semicircular canal; malformations; hearing loss
RESUMO
Os principais objetivos deste estudo observacional foram descrever uma malformação pouco reconhecida do ouvido interno designada malformação congénita bilateral dos canais semicirculares; determinar se o grau e padrão de dismorfia dos canais semicirculares e a presença ou ausência de uma cóclea bem formada são preditivos do resultado audiológico, tipo e gravidade da perda auditiva congénita; e investigar se esta malformação surge isoladamente ou em associação a formas sindrómicas já descritas. Foi realizada uma revisão de oito casos de perda auditiva com evidência radiológica de malformação congénita dos canais semicirculares. Foi recolhida informação sobre história clínica, exame físico, estudo imagiológico por tomografia computorizada e audiogramas seriados para todos os doentes. As características analisadas incluíram presença de outras dismorfias sugestivas de síndromes malformativas; avaliação da configuração, gravidade e tipo de perda auditiva nos exames audiométricos; tipo de reabilitação audiológica; e presença de outras anomalias do ouvido interno para além das alterações no sistema vestibular.
Entre os oito casos incluídos no estudo, seis apresentavam um síndrome ou anomalia cromossómica conhecida. A perda auditiva foi moderada a profunda em todos os casos. Todos os pacientes apresentavam deformidades dos canais semicirculares bilateralmente, com padrão anatómico geralmente idêntico em cada lado. Dos oito casos, seis apresentavam um desenvolvimento coclear normal; malformações na membrana timpânica e no canal auditivo externo foram encontradas apenas em um caso; alterações na cadeia ossicular foram descritas em três pacientes; o vestíbulo e aqueduto vestibular eram normais na maioria dos casos; anomalias no desenvolvimento da janela oval estavam presentes em dois casos.
O presente estudo mostra que é difícil estabelecer uma correlação entre a gravidade e o tipo de perda auditiva e as alterações radiográficas. A perda auditiva associada à displasia do canal semicircular é mais provável devido ao desenvolvimento anômalo do labirinto membranoso, que não é detetável radiologicamente pela tomografia computadorizada.
Palavras-chave: anomalias congénitas; canais semicirculares; malformações; perda auditiva
Introduction
The etiology of congenital hearing loss is variable. Congenital hearing loss can be an isolated finding or be part of a syndrome, with approximately one third of hearing losses being syndromic.1,2
The adult inner ear is divided into an auditory apparatus, consisting of the organ of Corti in the cochlea, and a vestibular apparatus, consisting of the saccule, utricle, and semicircular canals (SCCs). These structures are composed of a membranous labyrinth encased in a bony labyrinth, located in the petrous portion of the temporal bone.
The inner ear develops from an ectoderm thickening on the lateral surface of the neural tube termed otic placode. During the fourth week of human gestation, the otic placode invaginates into the underlying mesenchyme, forming the otic vesicle (otocyst). Development of the inner ear begins around the sixth week of fetal life, when the otic vesicle splits. The ventral part (pars inferior) becomes the vestibular sacculus and cochlear canal, which is fully formed by week nine; the dorsal part (pars superior) becomes the utricle and subsequently the SCCs at week eight; and the lateral canal is the last to form.3 It is generally agreed that inner ear deformities result from sudden developmental arrest during these successive stages, which is why the lateral canal is the most frequently involved.4 This malformation can be found as an isolated anomaly.
When not associated with hearing loss or acute acquired vertigo symptoms, isolated congenital abnormalities may not become clinically significant. Computed tomography (CT) can identify peripheral vestibular/cochlear lesions, especially anomalies such as deformities of the bony labyrinth. SCC anomalies are the most common congenital abnormalities of the inner ear, but their prevalence among inner ear malformations varies according to the overall severity of the anomaly.3,5
Materials and methods
Eight cases of bilateral congenital semicircular canal malformation (BCSCM) were retrospectively identified among patients evaluated for hearing loss. This was an observational study using a convenience sample of eight patients identified with the anomaly of interest. Cases were identified during routine clinical practice while performing CT scan in children evaluated for hearing loss. According to clinical practice, imaging study is performed to all children with mild-to-moderate or moderate-to-severe hearing loss, in cases of severe-to-profound deafness with negative genetic test, in children with unilateral hearing loss, and in children with conductive or mixed hearing loss.
Information on patient’s history, complete otologic examination, CT scan, and serial audiograms was retrieved for all patients and the type of audiological rehabilitation was described.
Among the eight identified cases, those associated with acknowledged syndromes had been previously identified in Pediatric/Genetic consultations.
All ears were evaluated for external auditory canal (EAC), tympanic membrane, ossicles, round and oval windows, cochlea, vestibule, SCCs (lateral, posterior, superior), vestibular aqueduct, mastoid and perigeniculate, and tympanic and mastoid segments of the facial nerve using one-millimeter-thin-section CT in both axial and coronal planes. For two cases, nuclear magnetic resonance (NMR) imaging evaluation was also available.
Cases were compared with those reported in the literature.
Results
Demographic findings, audiological evaluation, and type of rehabilitation of the eight cases are summarized in Table 1.
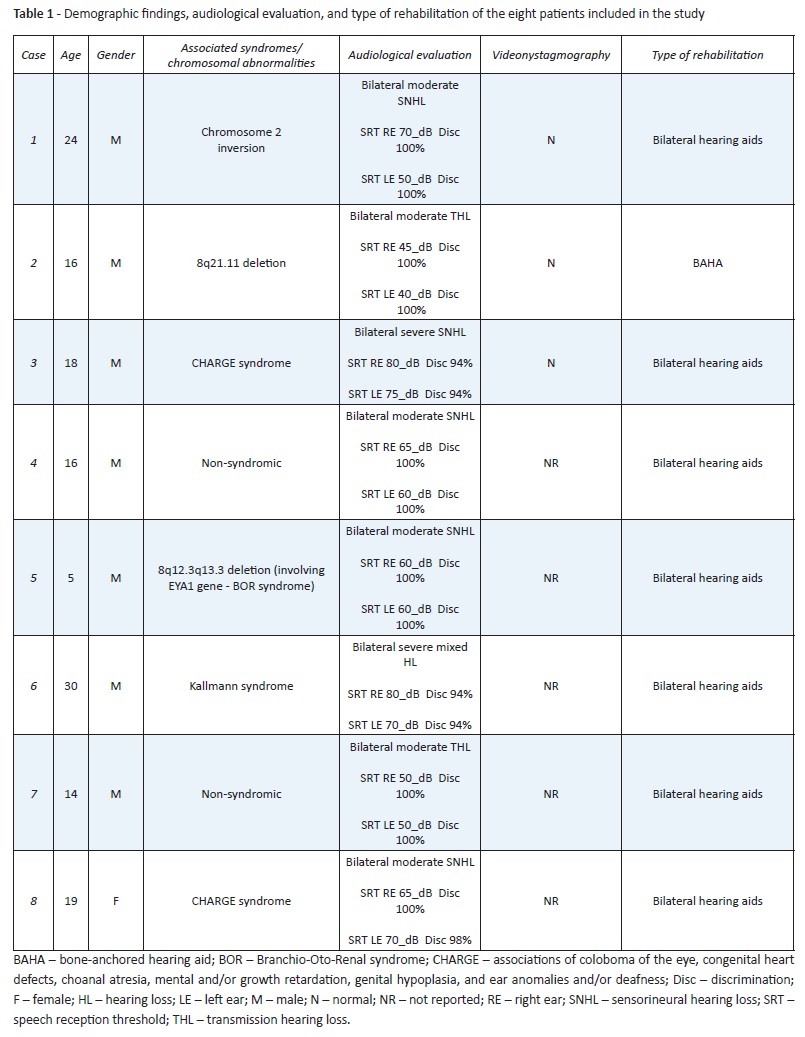
(clique para ampliar ! click to enlarge)
One female and seven male patients were identified with the study anomaly, with a mean age at the time of evaluation of 17.75 years.
Among the eight cases, two were initially believed to be non-syndromic/without chromosomal abnormalities, with the anomaly radiographically uncovered later during a hearing loss evaluation. Six patients had recognized syndromes/chromosomal abnormalities: two patients had been diagnosed with CHARGE association (coloboma, heart defects, atresia choanae, retardation of growth and development, genitourinary problems, and ear abnormalities); one patient had been diagnosed with Kallman syndrome (characterized by a rare hearing impairment presentation); and three patients had chromosomal abnormalities, including chromosome 2 inversion, 8q21.11 deletion, and 18q12.3q13.3 deletion (with involvement of EYA1 gene associated with brachiootorenal syndrome).
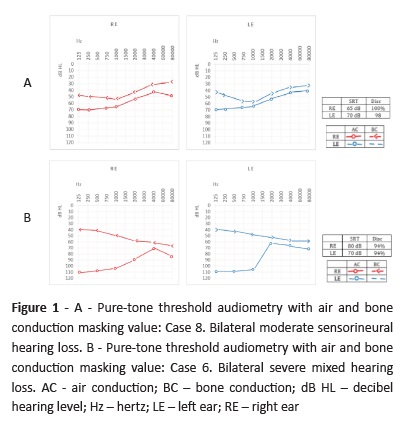
Hearing loss was moderate to profound in all cases, with similar degree and type of deafness on each side. Most patients (five in eight) had sensorineural hearing loss and two patients had transmission hearing loss (Cases 2 and 7). Only one patient had a mixed hearing loss (Case 6). Six cases clearly had more pronounced hearing loss between 250 and 1000 Hz.
Clinical history analysis enabled to investigate presence of vestibular symptoms. None of the patients had symptoms compatible with balance impairment. Three cases performed videonystagmography to clarify unspecific symptoms, with normal results.
CT findings regarding SCCs are shown in Table 2.
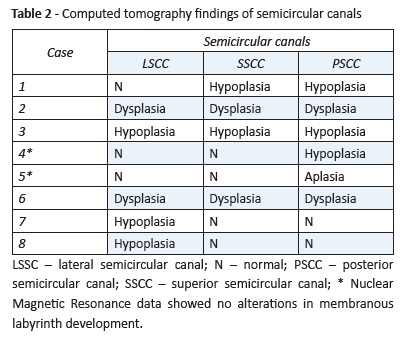
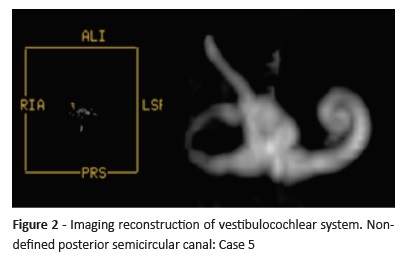
All patients had bilateral deformities, with usually identical anatomical pattern on each side. Three patients had hypoplasia (Case 3)/dysplasia (Cases 2 and 6) of all SCCs. Isolated posterior SCC hypoplasia was found in Case 4 and a probable total posterior SCC aplasia was found in Case 5. Isolated lateral SCC hypoplasia was found in two cases (Cases 7 and 8). Case 1 had stenosis/hypoplasia in the posterior and lateral SCC.
CT findings of the cochlea and vestibular structures are summarized in Table 3.
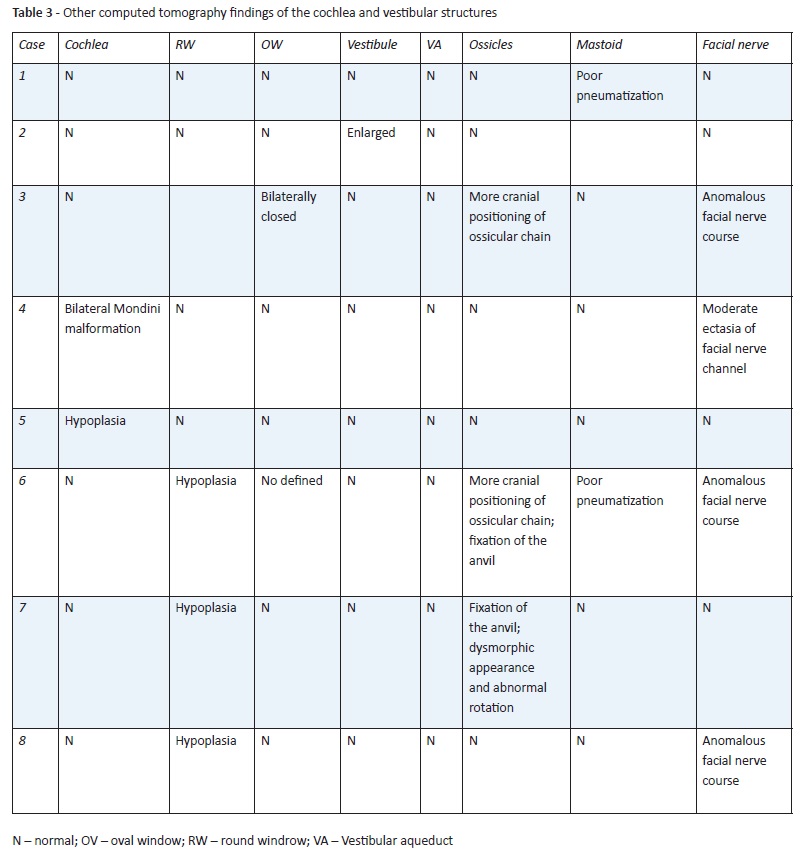
(clique para ampliar ! click to enlarge)
Of the eight cases, six had normal cochlear development, one had bilateral cochlear hypoplasia, and one had bilateral Mondini malformation. Malformations in the tympanic membrane and external auditory canal were only found in one case (Case 3). Changes in ossicular chain, such as fixation of the anvil, more cranial position, dysmorphic appearance, and abnormal rotation were found in three patients.
Vestibules and vestibular aqueduct were normal in most cases, except in Case 2, which had enlarged vestibules. Abnormalities of oval window development and hypoplasia were found in two cases (Cases 3 and 4), both syndromic. Poorly defined round window was found in three cases (Cases 6, 7, and 8). Development and pneumatization of the mastoid was quite poor in two cases. An anomalous course of the facial nerve was observed in four cases.
Seven of the eight patients used hearing aids, with good adaptation and auditory gain. One case had auditory rehabilitation with bone-anchored hearing aid (BAHA) (Case 2).
Discussion
Inner ear malformations with an impact on hearing status have been previously investigated. SCC malformations can be an isolated finding or be part of a syndrome. This anomaly seems to be very common among children with CHARGE association, who usually have at least a minor abnormality of the bony cochlea.5 In a series of 15 patients with SCC aplasia, Satar et al. found nine patients with CHARGE association.5 In the present case series, two had CHARGE association. In the study by Satar et al, four patients (approximately 27% of cases) were nonsyndromic.5 Similar results were observed in this series, in which only two cases (25%) were non-syndromic/without chromosomal abnormalities.
The unusual combination of Kallmann syndrome and deafness was observed in this study. Clinical suspicion of Kallmann syndrome is usually based on the combination of hypogonadism and anosmia/hyposmia and the association between Kallmann syndrome and hearing loss has been only sporadically reported.6,7
The 18q12.3q13.3 deletion with involvement of EYA1 gene associated with brachiootorenal syndrome was observed in Case 5. This represents a rare autosomal dominant disorder characterized by branchiogenic anomalies, hearing loss, and renal anomalies.8
Some authors argue that an inevitable result of SCC complete aplasia is a delay in walking and other locomotive activities in early childhood years, emphasizing the importance of SCCs in postural control acquisition.5 Delayed growth and development also complicate this situation in cases in which inner ear anomaly is related to CHARGE or other diseases with psychomotor retardation. However, isolated uni or bilateral SCC malformations can be clinically undetectable due to complete compensation.
Evaluation of inner ear malformations and their impact on balance was not the main objective of the present work. Although in this case series patients were older than five years and none presented clinical symptoms of imbalance or vertigo, clinical evaluation and patient collaboration for more specific diagnostic tests are hindered in cases of delayed psychomotor development.
Using the BIAP (Bureau International d’Audiophonologie) classification, hearing loss was classified as moderate (Pure-tone average of 60 dB) to severe (Pure-tone average of 80 dB) in all cases. Most patients (five in eight) had sensorineural hearing loss (severe hearing loss in Case 3 and moderate hearing loss in Cases 1, 4, 5, and 8). Patients with similar inner ear anomalies displayed different deafness types and degrees, showing how radiographic evaluation alone is insufficient to establish a correlation between severity and type of hearing loss. Hearing loss associated with SCC dysplasia is most likely due to anomalous membranous labyrinth development, which is not radiologically detectable by CT scan.1 The two cases in this study with NMR data showed no alterations in membranous labyrinth development. An interesting and not previously reported fact is that in the present case series six patients clearly presented a greater hearing loss at lower frequencies (between 250 and 1000 Hz).
Patients in this series exhibited a wide range of inner ear structural abnormalities. In general, the severity of any inner ear anomaly is believed to be dependent on the timing of the developmental insult. Considering the inner ear embryogenesis process previously described, dysplasia/hypoplasia of SCCs with a morphologically normal cochlea could not be easily explained by a very early nonspecific developmental insult, as the membranous vestibular labyrinth begins to develop earlier than the membranous cochlear duct.1,5 The high degree of variability in radiological findings and associated cochlear malformations argue against a single classification system based on embryogenic arrest.1,5 The advent of new molecular genetic analysis enables a much more complex history regarding the etiology and pathogenesis of SCC anomalies and suggests that cochlear and vestibular development may rely on independent, non-interacting mechanisms.
The lateral SCC is phylogenetically more recent, as demonstrated by jawless vertebrates exhibiting a two-canal inner ear composed of the anterior (superior) and posterior SCC.9 Among the SCC, lateral SCC malformations are the most frequent.10 In this study, isolated lateral SCC malformations were only observed in two cases.
Single malformation of the posterior SCC has been described in several diseases. Higashi et al. reported aplasia of the posterior SCC in 26% of cases associated with Waardenburg syndrome.12,13 Irie et al. described two cases of unilateral aplasia of the posterior SCC in Waardenburg syndrome.12,13 In the present study, isolated posterior SCC hypoplasia/aplasia was found in two cases, only one of which associated with chromosomal abnormalities.
Absence of SCCs has been reported in Goldenhaar syndrome and CHARGE association.14,15 In the present study, the three cases with alterations involving the three SCCs were all associated with syndromes/chromosomal abnormalities.
Six of the eight patients in this study had normal cochlear development. Cases with cochlea malformation did not have more severe hearing loss compared with the remaining, showing the absence of a specific pattern of audiological findings potentially associated with severity of cochlear radiographic findings. Patients with deformities preserving the cochlear architecture showed no consistently improved hearing compared with patients with cochlear malformations.
In this study, no association was found between malformations of round and oval windows, vestibule, ossicles, EAC, vestibular aqueduct, mastoid, or course of the facial nerve and severity of hearing loss. The described malformations were not more frequent in patients with recognized syndromes/chromosomal abnormalities. These findings do not agree with the results of Satar el al. showing remarkable differences in syndromic cases regarding the frequency of abnormalities of oval and round window development, unusual vascular abnotmalities within the temporal bone, and greater likelihood of anomalous facial nerve course.5
Conclusion
The present study shows that a correlation between the severity and type of hearing loss is difficult to establish based on analysis of radiographic abnormalities only. Hearing loss associated with SCC dysplasia is more likely due to anomalous membranous labyrinth development, which is not radiologically detectable by CT scan. Importantly, the small sample size in this study limits the power of results and conclusions. Studies with larger sample size and NMR analysis will certainly provide results with greater statistical power.
The evaluation of inner ear malformations and their impact on balance could be an interesting study to carry out in the future. Based on the complexity of inner ear development, it is reasonable to hypothesize that anomalies in this process are probably not just the result of arrested embryogenesis. Further studies with molecular analysis and high-resolution, three-dimensional reconstruction are necessary to better understand the complexity of inner ear malformations and development.
REFERENCES
1. Yu KK, Mukherji S, Carrasco V, Pillsbury HC, Shores CG. Molecular genetic advances in semicircular canal abnormalities and sensorineural hearing loss: a report of 16 cases. Otolaryngology-Head and Neck Surgery. 2003; 129:637-46. [ Links ]
2. Gettelfinger JD, John PD. Syndromic Hearing Loss: A Brief Review of Common Presentations and Genetics. Journal of Pediatric Genetics. 2018; 7:1-8. [ Links ]
3. Walther LE, Nath V, Krombach GA, Di Martino E. Bilateral posterior semicircular canal aplasia and atypical paroxysmal positional vertigo: a case report. Acta Otorhinolaryngologica Itall. 2008; 28:79. [ Links ]
4. Parnes LS, Chernoff WG. Bilateral semicircular canal aplasia with near-normal cochlear development: two case reports. Annals of Otology, Rhinology & Laryngology. 1990; 99:957-9. [ Links ]
5. Satar B, Mukherji SK, Telian SA. Congenital aplasia of the semicircular canals. Otology & neurotology. 2003; 24:437-46. [ Links ]
6. Salama N. Kallmann syndrome and deafness: an uncommon combination: A case report and a literature review. International Journal of Reproductive Biomedicine. 2016; 14:541. [ Links ]
7. Marlin S, Chantot-Bastaraud S, David A, Loundon N, Jonard L, Portnoï MF, et al. Discovery of a large deletion of KAL1 in 2 deaf brothers. Otol Neurotol. 2013; 34:1590-4. [ Links ]
8. Unzaki A, Morisada N, Nozu K, Ye MJ, Ito S, Matsunaga T, et al. Clinically diverse phenotypes and genotypes of patients with branchio-oto-renal syndrome. Journal of human genetics. 2018; 63:647. [ Links ]
9. Gavalas A, Studer M, Lumsden A, Rijli FM, Krumlauf R, Chambon P. Hoxa1 and Hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development. 1998; 125:1123-36. [ Links ]
10. Johnson J, lalwani AK. Sensorineural and conductive hearing loss associated with lateral semicircular canal malformation. Laryngoscope. 2000; 110:1673-9. [ Links ]
11. Higashi K, Matsuki C, Sarashina N. Aplasia of posterior semicircular canal in Waardenburg syndrome type II. J otolaryngol. 1992; 21:262-4. [ Links ]
12. Irie K, Ogata H, Mitsudome A. CT findings of the temporal bones in Waardenburg’s syndrome. No To Hattatsu. 1990; 22:241-6. [ Links ]
13. Xu GY , Hao QQ , Zhong LL, , Ren W , Yan Y , Liu RY , et al. SOX10 mutation is relevant to inner ear malformation in patients with Waardenburg syndrome. Zhonghua er bi yan hou tou jing wai ke za zhi= Chinese journal of otorhinolaryngology head and neck surgery. 2016; 51:832-7.
14. Choo DI, Tawfik KO, Martin DM, Raphael Y. Inner ear manifestations in CHARGE: Abnormalities, treatments, animal models, and progress toward treatments in auditory and vestibular structures. In American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 2017; 175:439-49. [ Links ]
15. Wineland A, Menezes MD, Shimony JS, Shinawi MS, Hullar TE, Hirose K. Prevalence of Semicircular Canal Hypoplasia in Patients With CHARGE Syndrome: 3C Syndrome. JAMA Otolaryngology-Head & Neck Surgery. 2017; 143:168-77. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Joana Raquel Costa
Department of Otolaryngology Head and Neck Surgery
Centro Hospitalar Universitário do Porto.
Largo do Prof. Abel Salazar
4099-001
Email: joana_cccosta@hotmail.com
Received for publication: 04.11.2018
Accepted in revised form: 24.05.2019

