










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.2 Porto jun. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i2.15607
CASE REPORTS | CASOS CLÍNICOS
A very rare cause of infantile spasms
Uma causa muito rara de espasmos infantis
Margarida Silva FonsecaI, Clara VieiraII, Rui ChorãoIII, Anabela BandeiraIV, Inês CarrilhoI
I. Department of Pediatric Neurology, Centro Materno-Infantil do Norte, Centro Hospitalar Universitário do Porto. 4050-371 Porto, Portugal. margarida_neils@hotmail.com; icccarrilho@gmail.com
II. Department of Pediatrics, Unidade de Vila Nova de Famalicão, Centro Hospitalar do Médio Ave. 4761-917 Vila Nova de Famalicão, Portugal. vieira.clara@gmail.com
III. Department Neurophysiology, Centro Hospitalar Universitário do Porto. rui.chorao@gmail.com
IV. Metabolic Diseases Unit, Department of Pediatric, Centro MaternoInfantil do Norte, Centro Hospitalar Universitário do Porto. 4050-371 Porto, Portugal. anabela.ol.bandeira@sapo.pt
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
Psychomotor development regression or delay associated with epilepsy represent a diagnostic challenge. The diagnostic approach should take into account age group, epileptic syndrome, physical and neurological data, and organ and/or system involvement. Herein is reported the case of a toddler for whom hair development, epileptic seizure evolution, and electroencephalographic findings were key for Menkes kinky hair disease diagnosis. The typical electroclinical evolution in this syndrome has rarely been previously reported.
A 22-month-old boy, born at 35 weeks, was admitted to the hospital by the age of two months due to epileptic seizures. Physical examination revealed dysmorphic facial features, pectus excavatum, and inguinal hernias. Antiepileptic drugs were initiated and one month later the patient was readmitted with recurrent epileptic seizures. Transfer to a hospital with Pediatric Neurology support was required, where light-toned and pleated skin, sparse hair, failure to thrive, and axial hypotonia were remarked. Initial investigation with general metabolic, neuroimaging, ophthalmological, and microarray study revealed no changes. Electroencephalograms were markedly abnormal, initially with focal changes and later with hypsarrhythmia. Considering the patient’s phenotype, copper serum level was analysed, with null value. Molecular study confirmed Menkes kinky hair disease and copper histidine therapy was initiated.
Menkes kinky hair disease should be considered in infants with global developmental delay, severe hypotonia, refractory epilepsy, and typical hair and skin changes occurring early in life. However, neonatal diagnosis is hampered by age-unspecific signs and symptoms. Despite being a rare and fatal entity, timely diagnosis allowing early therapy institution and avoiding unnecessary additional tests and prompt genetic counseling are of utmost importance.
Keywords: Copper transport disease; developmental disability; early diagnosis; electroencephalography; epilepsy; genetic counselling; hair; Menkes kinky hair disease; neurological manifestations
RESUMO
A regressão ou atraso do desenvolvimento psicomotor associados a epilepsia constituem um desafio diagnóstico. A abordagem diagnóstica deve considerar o grupo etário, a síndrome epilética, o exame físico e neurológico e o envolvimento de órgãos/sistemas. É aqui descrito o caso clínico de uma criança em que o aspeto do cabelo e a evolução das crises epiléticas e dos resultados eletroencefalográficos foram importantes para o diagnóstico de Doença de Menkes. A evolução eletroclínica típica desta síndrome foi raramente descrita noutras publicações.
Um rapaz de 22 meses, com 35 semanas de idade gestacional, foi admitido no hospital aos dois meses de idade devido a crises epiléticas. O exame físico revelou fácies dismórfico, pectus excavatum e hérnias inguinais. Foram iniciados antiepiléticos e um mês depois o rapaz foi readmitido por crises epiléticas recorrentes. O doente foi transferido para um hospital com apoio de Neurologia Pediátrica, onde se destacaram a pele clara e pregueada, cabelo escasso, má evolução ponderal e hipotonia axial. A investigação inicial através de estudo metabólico geral, neuroimagiológico, oftalmológico e de microarray não revelou alterações. Os eletroencefalogramas encontravam-se marcadamente anormais, inicialmente com alterações focais e posteriormente com hipsarritmia. Atendendo ao fenótipo do doente, foi efetuado o doseamento de cobre, com valor nulo. O estudo molecular confirmou o diagnóstico de Doença de Menkes e foi iniciada terapêutica com histidinato de cobre.
A Doença de Menkes deve ser considerada em lactentes com atraso global do desenvolvimento, hipotonia severa, epilepsia refratária e alterações típicas do cabelo e pele com instalação precoce, mas é de diagnóstico neonatal difícil devido a sinais e sintomas inespecíficos nesta idade. Apesar de ser uma entidade rara e fatal, o diagnóstico precoce para rápida instituição terapêutica, evicção de exames complementares desnecessários e aconselhamento genético atempado são de extrema importância.
A perda auditiva associada à displasia do canal semicircular é mais provável devido ao desenvolvimento anômalo do labirinto membranoso, que não é detetável radiologicamente pela tomografia computadorizada.
Palavras-chave: aconselhamento genético; alterações do desenvolvimento; cabelo; diagnóstico precoce; doença de Menkes; doença do transporte de cobre; eletroencefalografia; epilepsia; manifestações neurológicas
Introduction
Epileptic encephalopathies occur at Pediatric age and have a wide variety of etiologies and age-dependent specificities. When associated with developmental delay or regression, dysmorphisms, or other atypical findings on physical examination, diagnosis of inherited metabolism disorders and/or genetic syndromes should be considered.1 In neurometabolic disorders, epilepsy usually occurs at young ages (neonates or infants).1,2 Whenever typical or identifiable signs are present, they should guide clinical diagnosis.1-13
Herein is described the case of a patient for whom hair development was the key clue for Menkes kinky hair disease (MKHD) diagnosis, highlighting the importance of clinical features and electroencephalographic findings for establishing diagnosis.
Few reports are available in the literature reporting both typical phenotypic features and electrophysiological findings associated with MKHD.
Clinical case
Herein is presented the case of a 22-month-old boy admitted to a tertiary hospital with initial epileptic seizures at the age of two months.
Parents were apparently healthy and motor neuron disease was suspected in a second-degree relative (grandfather). Pregnancy and delivery were unremarkable, except for premature childbirth. The child was born at 35 weeks of gestation, with no need for resuscitation support. Fenton preterm growth chart percentiles were compatible with a small-for-gestational-age baby and he was admitted to the Neonatal Intensive Care Unit for 14 days, with no intercurrences. Physical examination revealed a particular triangular-shaped face, raised nostrils, prominent ears, and pectus excavatum. Lax skin and cephalohematoma were additionally registered.
On first hospital admission, the boy presented with focal left hemiclonic seizures. Epileptic seizures were observed and focal epileptiform activity was evident on electroencephalogram (EEG; Fig. 1 ). Treatment with sodium valproate (VPA) and phenobarbital (PB) was initiated and the patient was seizure-free on hospital discharge. Bilateral inguinal hernias and left cryptorchidism were detected on physical examination.
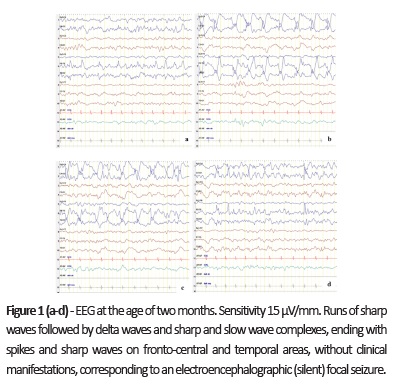
By the age of three and a half months, the boy was hospital readmitted with seizure recurrence and immediately transferred to a central hospital with Pediatric Neurology. VPA was withdrawn and switched to levetiracetam (LEV) in combination with previous PB, with clinical benefit. Thiamine supplementation was also started. Failure to thrive and global developmental delay were evident. On examination, dolichocephaly was noticed, together with axial hypotonia with brisk deep tendon reflex. Light-toned and pleated skin and steely and sparse hair were also observed.
Initial workup performed at the age of two months − infectious screening through institutional protocols; serum hematological and biochemical study, including specific metabolic study (lactate/pyruvate, uric acid, creatine kinase, cholesterol, total homocysteine, transferrin isoelectric focusing for carbohydrate-deficient glycoproteins, very long chain fatty acids, serum amino acids and urine organic acids); microarray study; brain magnetic resonance imaging (MRI); and ophthalmological evaluation − showed no changes. Attending to the patient’s phenotypic characteristics, metabolic study was complemented with copper and ceruplasmin quantification on second hospital admission, at the age of three and a half months. Serum values were 0 μmol/L and 0.070g/L, respectively. Molecular study for ATP7A gene was also conducted, disclosing ATP7A gene mutation c. 1639C>t (P.(ARG547*) and confirming MKHD diagnosis. Therapy with subcutaneous copper histidine (500 μg per day) was started, as well as regular follow-up in Pediatric Neurology and Metabolic Diseases outpatient clinic. After six months of therapy, copper and ceruplasmin levels were 9.27 μmol/L and 0.114 g/L, respectively. During this period, the patient was submitted to surgical repair of inguinal hernias, stopped thiamine supplementation, and maintained PB and LEV as antiepileptic drugs.
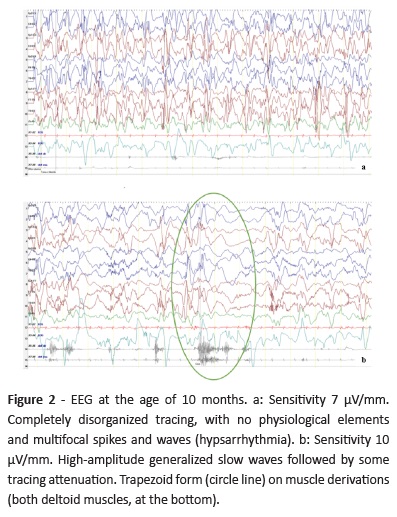
By the age of 10-11 months, the boy developed clusters of epileptic spasms. In EEG, disorganized tracing with multifocal high voltage sharp and slow wave abnormalities were evident, consistent with hypsarrhythmia (Fig. 2a), and epileptic spasms were recorded (Fig. 2b). Vigabatrin (VGB) was started, with epileptic spasm reduction but not complete remission, and LEV was withdrawn.
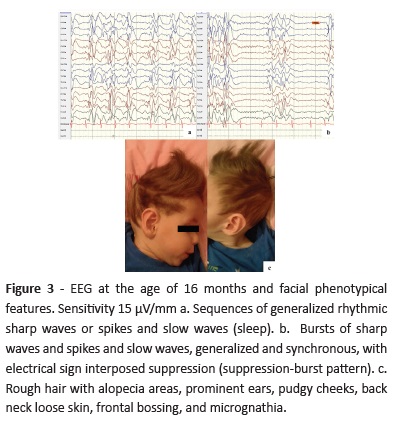
At 16 months of age, EEG showed multifocal epileptiform activity with burst-suppression pattern during sleep (Fig. 3) and rough hair with alopecia areas and other particular facial features were evident (Fig. 3c).
From initial follow-up until present, global developmental delay was remarkable, with poor visual contact and language regression (only guttural sounds), severe global hypotonia, and no postural control (the boy does not sit and has no head control). Global growth has been persistently below percentile 3 of World Health Organization Child Growth Standards. The patient has been on a soft adapted age diet, with caloric and nutritional supplementation (medium-chain triacylglycerols and maltodextrin complex carbohydrate). Currently, at 22 months, epilepsy became refractory, with recurrent epileptic spasms and tonic seizures despite antiepileptic drugs.
Maternal molecular study showed the same pathogenic gene variant.
Discussion/conclusions
MKHD diagnosis should be considered in infants with severe developmental delay, early-onset epilepsy (hemiclonic seizures or spasms), and peculiar hair appearance, as observed in the present patient. This rare disease entity has a 1/250,000 incidence and X-linked inheritance.3 It primarily affects males (1/140,000 male births), while females are usually unaffected carriers.3-7 The condition is characterized by generalized copper deficiency caused by mutations in ATP7A gene, which encodes a copper transport polypeptide-type ATPase that becomes defective.5,6,8 Besides MKHD most prevalent severe form, 9-15% of patients may have milder disease subtypes, such as ATP7A-related distal motor neuropathy (DMN) and occipital horn syndrome (OHS; the less severe form).1,3
The ATP7A protein is found throughout the body, except in liver cells.9 Copper uptake in the gastrointestinal tract and copper delivery to the brain are compromised, resulting in low copper blood levels and dysfunction of several copper-dependent enzymes. This causes elevated plasma lactate due to complex IV deficiency (cytochrome c oxidase), collagen crosslinking alteration (catalyzed by lysyl oxidase), and melanin production (tyrosinase), resulting in hair and skin abnormalities (pili torti, trichorrhexis nodosa, moniletrix, alopecia, light-toned hair and skin), vascular abnormalities (elongated vessels, tortuosity of intracranial vessels and subdural hematoma), and Purkinje cell degeneration in the cerebellum, as well as episodic hypothermia.1,3,4,6,9,10,13 Brain dopamine beta hydroxylase deficiency accounts for such pronounced neurological involvement.3 Neurological symptoms, like epileptic seizures and hypotonia, become evident at the second or third months of life as a result of cerebral neurodegeneration associated with such deficit.3,13
Due to unspecific early clinical signs - prematurity, large cephalohematomas, and skin laxicity −, neonatal diagnosis is challenging.3 All these features were present in this patient. Typical dermatological and neurological manifestations and developmental delay become evident later, as also observed in the present patient.
The typical sparse, steely, and brittle hair can appear at birth or be the first MKHD sign at the age of one/two months.1,3,4,6 In the present patient, hair distinctiveness was observed at the age of 3.5 months and was the main diagnostic clue, prompting further study.
Copper deficiency also implicates other body structures, like bone, and can manifest by osteoporosis, deformities, fractures, and exostosis.3,10
Failure to thrive, feeding difficulties, vomiting, and diarrhea represent additional MKHD symptoms. An odd appearance, frontal or occipital bossing, micrognathia, pudgy cheeks, pectus excavatum, and umbilical/inguinal hernias may also be observed.3,6 This patient exhibited most of these clinical features (Fig. 3c).
MKHD differential diagnosis includes Ehlers-Danlos syndrome, Marfan syndrome, cutis laxa, mitochondrial disorders, osteogenesis imperfecta, and child abuse.6,10
Initial diagnosis is based on clinical features (particularly typical hair changes) and supported by reduced levels of serum copper and ceruloplasmin. However, serum study alone in males under six months is problematic, given the normally low copper concentrations in infants this young.4 Definitive diagnosis is based on molecular genetic testing.4,6 A typical course of epilepsy has been reported in the literature, comprising an early stage with focal clonic seizures, often evolving into status epilepticus (at three months); an intermediate stage with epileptic spasms (at 6-11 months); and a late stage with multifocal seizures, tonic spasms, and myoclonus (at 20-25 months).12 This pattern was also observed in this patient, except that early epileptic seizures were not very frequent and status epilepticus was not observed.
Typical MRI may present with bilateral subdural hygromas, brain and cerebellar atrophy, white matter changes, and tortuous intracranial vessels.14,15 In the present case, MRI at the age of two months was normal and provided no diagnostic clue.
Overall, this patient fulfilled all MKHD diagnostic criteria: clinical features, EEG pattern, serum biomarkers, and molecular identification of ATP7A gene mutation.
Genetic counseling is extremely relevant in this context, as MKHD has an X-linked recessive inheritance. Approximately one third of affected males has no family history of MKHD/OHS/DMN. However, if the mother is heterozygous there is a 50% risk of transmitting ATP7A pathogenic variant in each pregnancy.4 In this study, maternal molecular study showed the same pathogenic variant.
In the most prevalent and severe MKHD form, the vast majority of patients dies before the age of three years, usually from infections or vascular complications.3 Early copper histidine treatment start within the first postnatal weeks extends survival beyond the age of three. Medical care including feeding techniques can also potentially improve the very poor MKHD prognosis.3,7 In this case, besides adapted diet and caloric supplementation, the patient did not require further feeding support.
MKHD requires early diagnosis for prompt treatment start and multidisciplinary medical and nutritional surveillance to enable the most extended and quality lifetime. Additionally, timely diagnosis is key to avoid redundant studies and encourage adequate genetic counseling.
REFERENCES
1. Hoffmann GF, Zschocke J, Nyhan WL. Inherited Metabolic Diseases. Berlin: Springer; 2017. [ Links ]
2. Karimzadeh P. Approach to Neurometabolic Diseases from a Pediatric Neurological Point of View. Iranian Journal of Child Neurology. 2015; 9:1-16. [ Links ]
3. Bierings M, Clayton P, Houwen R. Disorders of Metal Transport (2012) In Saudubray JM, Berghe G, Walter JH eds. Inborn Metabolic Diseases: Diagnosis and Treatment. Berlin: Springer; 2012. 539-40p. [ Links ]
4. Kaler SG. ATP7A-Related Copper Transport Disorders. 2003 May 9 [cited 2018 Mar 1]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1413/. [ Links ]
5. Online Mendelian Inheritance in Man (OMIM). URL: http://omim.org/entry/309400. Consulted on 02-04-2018. [ Links ]
6. Orphanet.URL: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=56. Consulted on 02-04-2018. [ Links ]
7. Kodama H, Fujisawa C, Bhadhprasit W. Inherited Copper Transport Disorders: Biochemical Mechanisms, Diagnosis, and Treatment. Curr Drug Metab. 2012; 13:237-250. [ Links ]
8. Tümer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013; 34:417-429. [ Links ]
9. Genetics Home Reference. URL: https://ghr.nlm.nih.gov/gene/ATP7A#resources. Consulted on 01-03-2018. [ Links ]
10. Kaler SG. Inborn errors of copper metabolism. Handb Clin Neurol. 2013; 113:1745-54. [ Links ]
11. Marques JS, Mateus M, Torres T, Santos H, Real MV, Santos F. Visual diagnosis: 8-day-old hypotonic newborn with sparse hair. Pediatr Rev. 2014; 35:e53-e56. [ Links ]
12. Bahi-Buisson N, Kaminska A, Nabbout R, Barnerias C, Desguerre I, De Lonlay P, et al. Epilepsy in Menkes disease: analysis of clinical stages. Epilepsia. 2006; 47:380-6. [ Links ]
13. Campistol J, Plecko B. Treatable newborn and infant seizures due to inborn errors of metabolism. Epileptic Disord. 2015; 17:229-42. [ Links ]
14. Manara R, D’Agata L, Rocco MC, Cusmai R, Freri E, Pinelli L. et al. Neuroimaging Changes in Menkes Disease, Part 1. Am J Neuroradiol. 2017; 38:1850 -57. [ Links ]
15. Manara R, Rocco MC, D’Agata L, Cusmai R, Freri E, Giordano L. et al. Neuroimaging Changes in Menkes Disease, Part 2. Am J Neuroradiol. 2017; 38:1858-65. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Margarida Silva Fonseca
Department of Pediatric Neurology
Centro Materno-Infantil do Norte
Centro Hospitalar Universitário do Porto
Largo da Maternidade
4050-371 Porto, Portugal
Email: margarida_neils@hotmail.com
Acknowledgements
The authors acknowledge the patient and family for study collaboration and consent.
Received for publication: 18.11.2018. Accepted in revised form: 25.06.2019

