










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.2 Porto jun. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i2.15091
CASE REPORTS | CASOS CLÍNICOS
Eating disorder - A diagnosis of exclusion
Perturbação do comportamento alimentar - Um diagnóstico de exclusão
Margarida Silva FonsecaI, Helena SantosII, Raquel GuedesI, Hugo Braga TavaresI
I. Adolescent Medicine Unit, Department of Pediatrics, Centro Hospitalar Vila Nova de Gaia/Espinho. 4400-129 Vila Nova de Gaia, Portugal. margarida_neils@hotmail.com; raquel.guedes@chvng.min-saude.pt; hugo.tavares@chvng.min-saude.pt
II. Infancy and Adolescent Neurosciences Unit, Department of Pediatrics, Hospitalar Vila Nova de Gaia/Espinho. 4400-129 Vila Nova de Gaia, Portugal. maria.helena.santos@chvng.min-saude.pt
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
In adolescents with weight loss, diagnoses other than eating disorders should be considered, namely neurological diseases. A 16-year-old girl with an intellectual development disorder was referred to the Adolescent Medicine outpatient clinic from Child Psychiatry with a diagnosis of eating disorder and persistent anemia. Her body mass index was consistently below the fifth percentile and long-lasting eating difficulties were reported since the age of 15. The girl had no other gastrointestinal, articular, or respiratory complaints, neither polyuria, polydipsia, or recurrent fever. Parental divorce and domestic violence were reported. The patient complained of excessive daytime sleepiness, asthenia, intermittent myalgia, and muscular weakness episodes. Phenotypic characteristics and personal medical history led to clinical suspicion of a neuromuscular disease and genetic study confirmed myotonic dystrophy type 1. This case highlights the importance of considering other diagnoses besides eating disorders in adolescents with eating problems. An exhaustive evaluation of personal and family medical history, patient complaints, and detailed physical examination is mandatory.
Keywords: adolescence; eating disorders; genetic testing; myotonic dystrophy type 1
RESUMO
Em adolescentes com défice de ganho ponderal, devem ser considerados outros diagnósticos para além de perturbações do comportamento alimentar, nomeadamente doenças neurológicas. Uma adolescente de 16 anos de idade com perturbação do desenvolvimento intelectual foi referenciada da consulta de Pedopsiquiatria para a de Medicina do Adolescente com diagnóstico de perturbação do comportamento alimentar e anemia persistente. Apresentava um índice de massa corporal consistentemente abaixo do percentil cinco e dificuldades alimentares desde os 15 anos de idade, sem outras queixas gastrointestinais, articulares ou respiratórias e sem poliúria, polidipsia ou episódios de febre recorrente. Foi reportado divórcio parental e violência doméstica. A rapariga referia sonolência diurna excessiva, astenia e episódios de mialgia intermitente e fraqueza muscular. O fenótipo e a história médica pessoal levaram à suspeita clínica de doença neuromuscular e o estudo genético confirmou o diagnóstico de distrofia miotónica tipo 1. Este caso realça a importância de considerar outros diagnósticos para além das perturbações do comportamento alimentar em adolescentes com problemas alimentares/de peso. Uma avaliação exaustiva da história médica pessoal e familiar e das queixas reportadas pelo doente e um exame físico detalhado são mandatórios.
Palavras-chave: adolescência; distrofia miotónica do tipo 1; estudo genético; perturbações do comportamento alimentar
Introduction
Eating disorders (EDs) are complex conditions with profound psychosocial and physical consequences, including high mortality rate. Despite growing recognition of their prevalence and severity, EDs remain underdiagnosed and even misdiagnosed.1 Recognition of ED-characteristic clinical presentation is key to distinguish these conditions from other important diseases with avoidance/restrictive food intake patterns. Pediatricians play an important role in correctly identifying these patients, avoiding inadequate treatment and properly managing the disease.
Myotonic dystrophy type 1 (DM1, Steinert’s disease) is characterized by autosomal dominant progressive myopathy, muscle weakness, and myotonia with multiorgan involvement. It results from a CTG trinucleotide repeat expansion in the 3’-untranslated region of the DMPK gene.2 Pathophysiology appears to involve RNA toxicity resulting from the expanded repeat in mutant DM1 allele transcripts.3 In adulthood, Steinert’s disease is the most frequent muscular dystrophy.4
DM1 phenotypical characteristics can be divided into muscular and extra-muscular. Muscular features include weakness of facial, neck, forearm, hand intrinsic, and foot dorsiflexor muscles, clinical myotonia and muscle pain, respiratory muscle affection, dysphagia, and dysarthria, which can all contribute to pneumonia or later respiratory failure and mastication and trouble eating.2,5-6 Extra-muscular features include cataracts, progressive cardiomyopathy and cardiac conduction disturbances (commonly late in the clinical course), cognitive impairment and adulthood personality disturbance, endocrine disturbances (like glucose intolerance and hypogonadism), and gastrointestinal disorders, like irritable bowel symptoms and gall stones.2,7
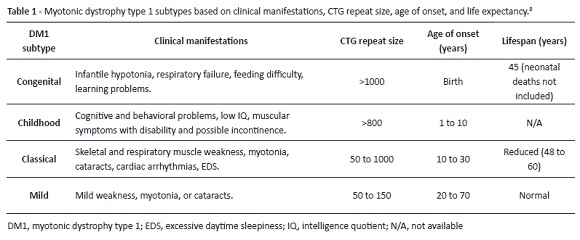
A systematic DM1 approach should distinguish between four disease subtypes: congenital, childhood, classic, and mild (Table 1). Each subtype severity correlates with CTG repeat expansion, despite considerable variability and overlap between DM1 forms.8 Asymptomatic patients are premutation carriers with a CTG repeat size between 38 and 49.2
Muscular problems (dysphagia, mastication difficulty) affecting head and neck in DM1 may result in particular eating behaviors, which can mislead ED diagnosis, as illustrated in the present clinical case.
Clinical case
A 16-year-old girl was referred to the Adolescent Medicine outpatient clinic (AMC) from Child Psychiatry due to persistent iron deficiency anemia. She was diagnosed at the age of 15 years with a restrictive-type ED, with depressive behavior and iron deficiency anemia. The girl had been treated with sertraline (50 mg daily) for 18 months and oral iron (III)-polymaltose (375 mg daily) for 16 months. She was also followed by a psychologist in primary care setting and by a dietitian in hospital setting, as part of ED multidisciplinary approach.
Patient’s prenatal history comprised premature threatening labor at 28 weeks of gestation, chronic maternal hypertension, and gestational diabetes without need for insulin therapy. No perinatal or neonatal health issues were reported.
Due to learning disabilities and expressive language difficulties, the girl was referred to the Pediatric Neurodevelopmental consultation at the age of 8. She was tested and revealed a Wechsler Intelligence Scale for Children (WISC) intelligence quotient score of 50 with limitations in the practical component of adaptive functioning, being diagnosed with moderate Intellectual Developmental Disorder of undetermined etiology (normal karyotype and fragile-X testing). Additionally, attention deficit hyperactivity disorder (ADHD) was also diagnosed and methylphenidate was started.
Progressive failure to thrive with no stature impact (50th percentile) was noted from 6 years old, with weight curves crossing from the 50th to the 10th percentile at 12 years and to below the 5th percentile at 15 years. Family described bizarre eating behaviors at the time of school initiation (6 years old), including solid food rejection, food hiding, easy choking on food, and restrictive/selective food preferences. These issues were interpreted as secondary to social stress by the general practitioner (GP), who had no clinical suspicion of ED at the time, and also disregarded by the girl’s family.
Family dysfunction characterized by domestic violence and alcohol abuse resulted in parental divorce at the age of 15 years.
At 8 years old, the girl began mentioning muscular pain at rest (back, shoulders, arms, and lower legs), exercise intolerance/muscle weakness, easy fatigue, and excessive day sleepiness. Sleeping with eyes open and mouth breathing were also reported by the family, but these signs were again not properly considered by the GP and no study was conducted.
The girl’s mother had a history of obesity, cataracts, depression, and nephrolithiasis. Maternal second and third-degree relatives were also reported to have cataracts, nephrolithiasis, and cardiomyopathy. An unclear history of a paternal second-degree relative with muscular weakness and/or bones disease was mentioned, but this information could not be confirmed. The patient had one only sister with an infant son, both apparently healthy.
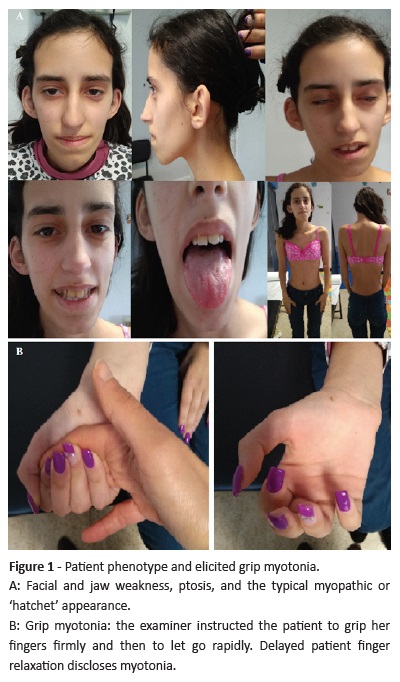
In AMC, at the age of 16, the girl had a body mass index (BMI) below the 5th percentile. Physical examination revealed thinness, skin pallor, nasal tone voice, facial muscle weakness with characteristic appearance − ptosis, long and narrow face, hollowed cheeks, and jaw sags (Figure 1A) −, clinical myotonia (Figure 1B), and hand intrinsic muscle weakness (compromised finger dexterity). Menarche occurred at 10 years old and the girl had a 6-month oligomenorrhea period. Puberty assessment revealed Tanner stage 5 for breast and pubic hair development. She maintained iron deficiency anemia diagnosed at 14 years old, when oral iron supplementation was started. Hypogonadotropic hypogonadism (luteinizing hormone and follicle-stimulating hormone 0.1 IU/L and estradiol inferior to 18.4 pmol/L) with secondary amenorrhea was diagnosed. A combined oral contraceptive (COC) was started due to menstrual irregularities and sexual risk.
Abnormal eating behavior persisted and included time-consuming and forced meals, deprecated food, and food hiding. Considering the girl’s complaints (namely excessive daytime sleepiness, asthenia, and intermittent myalgia episodes), physical exam findings, and family history pattern, diagnosis of a muscular condition was considered. DMPK gene triplet repeat analysis revealed one normal allele and the other with 800-900 trinucleotide (CTG) repeats, confirming myotonic dystrophy type 1 (DM1).
A workup plan for comorbidity evaluation was started based on a multidisciplinary evaluation including Cardiology (to exclude conduction abnormalities and myocardial dysfunction), Ophthalmology (to exclude cataracts), Gynecology (to monitor the best contraceptive method), Respiratory/Nose, Throat, and Ear evaluation (to monitor pulmonary function and sleep pattern/disturbance), Endocrinology (to exclude glucose and thyroid dysfunction), and Nutrition (to establish adequate diet and nutritional rehabilitation).
The patient is currently overall functional, despite some autonomy issues regarding daily life activities and self-care. She has good communicative skills and recently started a professional degree course.
Discussion/conclusion
Considering the patient’s clinical evolution (age of symptom onset, muscular and cognitive impairment) and DMPK gene triplet number quantification (800 CTG), childhood is the most likely DM1 subtype in this case.
Notwithstanding DM1 clinical suspicion, genetic testing for DMPK expanded CTG repeat is the diagnostic gold standard. Although not always present in younger ages, suggestive myotonic discharge may be detected by electromyography as the child ages.2
Despite presenting with suggestive physical and neuropsychological signs and symptoms since the age of 6, this patient was never investigated for neuromuscular conditions, and the persistent feeding and behavior problems were always labeled as ED.
There is currently no disease-modifying therapy available for DM1, although potential genetic treatments seem promising in a near future.2,8 Disease prognosis (life expectancy) depends on DM1 subtype (Table 1), but multidisciplinary follow-up and close surveillance for possible clinical complications seem to be of upmost importance. Annual electrocardiograms should be part of routine management.2
Genetic counseling is mandatory due to the 50% possibility of offspring disease inheritance (usually with increased CTG repeat number, disease severity, and age of onset anticipation), allowing a timely approach to potential comorbidities.2
This case highlights the need for a high index of clinical suspicion for diagnosing specific conditions through anamnesis, clinical exam, and multidisciplinary approach.
Although this neurological disorder could have been easily overseen in early development stages, it became increasingly evident throughout the years and its recognition could have prevented misdiagnosis and unnecessary treatments. Time from symptom onset, prolonged clinical course, absence of growth affectation (height and puberty), and presence of physical particularities should have triggered DM1 diagnosis at an earlier stage.
COCs are not indicated in amenorrheic adolescent girls with ED, as they can be associated with body changes.9,10 Regarding patient-related concerns (described above), earlier and correct diagnosis could have allowed to propose other (non-hormonal) contraceptive methods.
Family complaints, present and past medical history − including learning disorders, feeding history, and any related issues −, performance in daily routine activities, and a detailed physical and neurological medical examination should be part of patient approach.
REFERENCES
1. Rome ES, Strandjord SE. Eating Disorders. Pediatric in Review. 2016; 37:323-336. [ Links ]
2. Meola G, Cardani R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta. 2015; 1852:594-606. [ Links ]
3. Fiszer A, Krzyzosiak WJ. RNA toxicity in polyglutamine disorders: concepts, models, and progress of research. J Mol Med. 2013; 91:683-91. [ Links ]
4. Bouchard JP, Cossette L, Bassez G, Puymirat J. Natural history of skeletal muscle involvement in myotonic dystrophy type 1: a retrospective study in 204 cases. J Neurol. 2015; 262:285-93. [ Links ]
5. Engvall M, Sjögreen L, Kjellberg H, Robertson A, Sundell S, Kiliaridis S. Oral health status in a group of children and adolescents with myotonic dystrophy type 1 over a 4-year period. Int J Paediatr Dent. 2009; 19:412-22. [ Links ]
6. Umemoto G, Nakamura H, Oya Y, Kikuta T. Masticatory dysfunction in patients with myotonic dystrophy (type 1): a 5-year follow-up. Spec Care Dentist. 2009; 29:210-4. [ Links ]
7. Lau JK, Sy RW, Corbett A, Kritharides L. Myotonic dystrophy and the heart: A systematic review of evaluation and management. Int J Cardiol. 2015; 184:600-8. [ Links ]
8. Turner C, Hilton-Jones D. Myotonic dystrophy: diagnosis, management and new therapies. Curr Opin Neurol. 2014; 27:599. [ Links ]
9. Hicks CW, DeMarsh S, Singh H, Gillespie LA, Worley S, Rome ES, et al. Knowledge about various contraceptive methods in young women with and without eating disorders. Int J Eat Disord. 2013; 46:171-6. [ Links ]
10. Bergström I, Crisby M, Engström AM, Hölcke M, Fored M, Kruse PJ, et al. Women with anorexia nervosa should not be treated with estrogen or birth control pills in a bone-sparing effect. Acta Obstet Gynecol Scand. 2013; 92:877-80. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Margarida Silva Fonseca
Adolescent Medicine Unit
Department of Pediatrics
Centro Hospitalar Vila Nova de Gaia/Espinho
Rua Doutor Francisco Sá Carneiro
4400-129 Vila Nova de Gaia
Email: margarida_neils@hotmail.com
Acknowledgements
The authors acknowledge the patient and her family for study collaboration and consent.
Receive for publication: 24.09.2018. Accepted in revised form: 24.09.2019

