










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.2 Porto jun. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i2.15113
CASE REPORTS | CASOS CLÍNICOS
Hidden by the hair - A precocious puberty case report
Escondido pelo cabelo - Um caso de puberdade precoce
Filipa A. FerreiraI, Sara T. CostaI, Carla PereiraII, Brígida RobaloII, Lurdes SampaioII
I. Department of Pediatrics, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte. 1649-028 Lisboa, Portugal. filipafonsoferreira@gmail.com; sara.tbferreiracosta@gmail.com
II. Pediatric Endocrinology Unit, Department of Pediatrics, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte. 1649-028 Lisboa, Portugal. carlapereira@netcabo.pt; brigidarobalo@sapo.pt; mlurdesampaio@gmail.com
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
McCune-Albright syndrome (MAS) is one of the conditions causing precocious gonadotropin-independent puberty. It is a rare disease, characterized by two of the three following features: precocious puberty, polyostotic fibrous dysplasia (PFD), and café au lait (CAL) skin spots. Herein is presented the case of a girl with pubarche, acne, and transient thelarche since the age of three and menarche since the age of six years old. Besides transitory follicular cysts and advanced bone age, no other changes were found. Diagnosis was only established after brain magnetic resonance imaging showed fibrous dysplasia involving left craniofacial bones. The girl’s parents later mentioned that she had multiple café au lait skin spots on the scalp since birth, disclosing how the key diagnostic sign had been covered by the child’s hair.
MAS is a rare disorder and diagnosis depends on a high index of suspicion. CAL skin spots are generally the first manifestation, but can easily go unnoticed. Additionally, PFD may only affect some bones, like craniofacial.
Keywords: café au lait skin spots; dysplasia; McCune-Albright syndrome; polyostotic fibrous; precocious puberty
RESUMO
A puberdade precoce periférica pode ter várias etiologias, entre as quais a síndrome McCune-Albright (SMA). Trata-se de uma doença rara, definida por um mínimo de dois de três critérios clínicos: puberdade precoce, displasia fibrosa poliostótica (DFP) e manchas café-au-lait (CAL). Apresenta-se o caso de uma criança do sexo feminino com pubarca, acne e telarca intermitente desde os três anos de idade e menarca desde os seis. Dos exames complementares destacavam-se apenas alguns quistos foliculares transitórios e um avanço de idade óssea. O diagnóstico foi possível por ressonância magnética crânio-encefálica, que evidenciou a existência de DFP envolvendo os ossos do crânio e hemiface esquerda. Apenas mais tarde foi referido pelos pais que a criança tinha múltiplas manchas CAL no couro cabeludo desde o nascimento, evidenciando como neste caso a chave para o diagnóstico se encontrava escondida pelo cabelo.
A SMA é uma doença rara cujo diagnóstico depende de um elevado índice de suspeição. As manchas CAL geralmente são as primeiras manifestações, mas podem facilmente passar despercebidas. A DFP pode afetar apenas alguns ossos, como os craniofaciais.
Palavras-chave: displasia fibrosa poliostótica; manchas café au lait; puberdade precoce; síndrome McCune-Albright
Case report
A three-year-old girl was referred to our Pediatric Endocrinology Unit due to pubic hair growth since the age of 30 months. On clinical examination, she was on the 75th weight and 50th height percentile (according to WHO criteria), Tanner stage A1/P2/B1, and had two hyperpigmented skin lesions (6x10 cm on the back; 2x5 cm in the neck). No other findings were noticed. Laboratory study indicated elevated S-DHEA (192 μg/dL), normal total testosterone (13.9 ng/dL), normal TSH/FT4 (2.11 μU/mL; 0.93 ng/dL), and normal cortisol (9.1 μg/dL), IGF-1 (102 ng/mL), and prolactin (5.2 ng/mL). Adrenocorticotropic hormone (ACTH) test was normal and basal luteinizing hormone (LH), follicle-stimulating hormone (FSH), and estradiol levels were also normal (2.72 U/L; <0.07 U/L; <19.0 pg/mL). At the age of four, the child had facial acne, more pubic hair (Tanner P3), and advanced bone age of more than two years. Six months later, bilateral mammary gland was palpable (Tanner B2P3), although intermittently with posterior regression, and pelvic ultrasound revealed a 27-mm simple cyst on the right ovary and a 6.2-mm cyst on the left ovary. A few days later, gonadotropin-releasing hormone (GnRH) stimulation test was performed, but LH, FSH, and estradiol were on pre-pubertal levels. One year later, the girl had grown over 8 cm/year (>97th percentile of height velocity) and mammary gland was again not palpable (Tanner A2/P3/B1). First menstrual bleeding appeared almost two years later when the girl was six years and three months old. Subsequently, GnRH test showed elevated estradiol with low LH/FSH, compatible with peripheral precocious puberty. Despite this, considering the dramatic puberty evolution and to completely rule out a central precocious puberty cause, brain magnetic resonance imaging (MRI) was conducted, revealing signs of polyostotic fibrous dysplasia of the skull base and left temporal, parietal, and facial bones. Diagnosis of McCune Albright syndrome (MAS) presenting with precocious puberty and polyostotic fibrous dysplasia was considered at this point. Only later did the parents mention multiple scalp lesions (corresponding to café au lait [CAL] skin spots) present on the girl’s scalp since birth and not easily visible.
A luteinizing hormone-releasing hormone (LHRH) analog was initiated (monthly, via intramuscular administration) to suppress the secondary central precocious puberty that ultimately occurred.
Child’s wellbeing currently remains unaltered. She is on the 75th height percentile, has no additional organ involvement, and attends follow-up consultations twice a year.
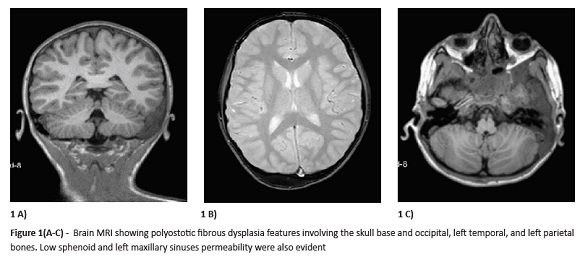
Discussion
MAS is a rare sporadic disease with an estimated prevalence between 1/100,000 and 1/1,000.000.1 It is caused by a missense somatic mutation in the α-subunit-encoding gene of G-protein, leading to activation of several hormonal receptors, as ACTH, TSH, FSH, and LH. It is acknowledged as a postzygotic mutation and therefore each patient presents somatic mosaicism.2 By definition, MAS is characterized by two of three clinical features: precocious puberty, polyostotic fibrous dysplasia, and café au lait skin spots. However, since virtually every gland or tissue can be affected, several clinical phenotypes are possible. Although precocious puberty is by far the most frequent, other hyperfunctional autonomous endocrinopathies may also be present: hyperthyroidism, Cushing’s syndrome, growth hormone (GH) excess, and renal phosphate wasting with or without rickets/osteomalacia. Other organs may also be involved, as the liver, heart, pancreas, or those of the gastrointestinal system, but represent less common features.3,4 It should be noticed that most clinical signs are present on first examination and generally persist throughout life (except for Cushing’s syndrome, phosphaturia, and precocious puberty). It is therefore crucial to be aware of all possible manifestations to make a diagnosis.4
Diagnosis is usually established based on clinical manifestations, but several laboratory and imaging tests may be useful. Skeletal radiograph may evidence the “ground glass” appearance of PFD bones; pelvic ultrasound may reveal cyst or ovary enlargement; biochemical findings may disclose high estradiol and low FSH/LH levels (compatible with peripheral precocious puberty) or other alterations (TSH/T4, GH, cortisol), depending on which endocrinopathy occurs.2
Other precocious puberty causes (such as congenital adrenal hyperplasia, brain lesions, ovarian tumors) and CAL skin spots (neurofibromatosis type 1) should be considered in the differential diagnosis.
Since there is no specific MAS treatment, therapeutic measures are directed at (developing) signs and symptoms. In the present case, precocious puberty was pharmacologically treated due to the potential psychological and final stature consequences. Data regarding the use of bisphosphonates − primarily used for bone pain reduction − in PFD is conflicting.5,6 Nonetheless, this patient did not present such symptom.
Although MAS is generally not associated with increased mortality and morbidity, several factors should be assessed: (i) which (and when) endocrine disorders arise, (ii) other solid organ involvement, presence of (iii) fractures or (iv) malignancies, among others. Being a multisystemic condition, disease management is challenging and should be conducted in a specialized center.5
Conclusion
This was a challenging case, as most diagnostic clues were hidden by the patient’s hair. Initial evaluation was based on common precocious puberty causes, but transient physical signs (telarche and ovarian cysts) plus normal hormone levels and absence of other signs hampered their interpretation.
Take-home messages:
• Although rare, MAS should always be considered a precocious puberty etiology.
• CAL skin pigmentation should raise clinical suspicion of MAS.
• MAS diagnosis is challenging and may require careful full body examination and several endocrine tests.
• There is no cure for MAS and treatment options vary according to clinical manifestations. Treatment goal (whether medical or surgical) is to relieve symptoms and offset physical shortcomings, ensuring the best possible quality of life.
REFERENCES
1. Raus I, Coroiu RE. McCune Albright syndrome - association of fibrous dysplasia, café-au-lait skin spots and hyperthyroidism - case report. Clujul Med [Internet]. 2016; 89:559. [ Links ]
2. Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis [Internet]. 2008; 3:12. [ Links ]
3. Lourenco R, Dias P, Gouveia R, Sousa AB, Oliveira G. Neonatal McCune-Albright syndrome with systemic involvement: a case report. J Med Case Rep [Internet]. 2015; 9:189. [ Links ]
4. Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis [Internet]. 2012; 7:S4. [ Links ]
5. Shaw N. McCune Albright Syndrome CR. ESPE Summer Sch. 2016;1-18. [ Links ]
6. Aragão ALA, Silva IN. Oral Alendronate Treatment for Severe Polyostotic Fibrous Dysplasia due to McCune-Albright Syndrome in a Child: A Case Report. Int J Pediatr Endocrinol [Internet]. 2010; 2010:432060. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Filipa A. Ferreira
Department of Pediatrics
Hospital de Santa Maria
Centro Hospitalar Universitário Lisboa Norte
Av. Prof. Egas Moniz
1649-028 Lisboa, Portugal.
Email: filipafonsoferreira@gmail.com
Received for publication: 28.09.2018. Accepted in revised form: 07.10.2019

