










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.3 Porto set. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i3.14261
CASE REPORTS | CASOS CLÍNICOS
Juvenile dermatomyositis: common manifestations of a rare disease
Dermatomiosite juvenil: manifestações comuns de uma doença rara
Mariana CapelaI, Joana ReisI, Diana SoaresI, Andreia RibeiroI, Isabel Pinto PaisII, Lúcia Trindade RodriguesIII
I Department of Pediatrics, Centro Hospitalar Vila Nova de Gaia/Espinho. 4400-129 Vila Nova de Gaia, Portugal. marianacarvalhocapela@gmail.com; joanasoaresdosreis@gmail.com; dianarsoares@gmail.com; andreia.ribeiru@gmail.com
II Department of Pediatric Gastroenterology, Centro Hospitalar Vila Nova de Gaia/Espinho. 4400-129 Vila Nova de Gaia, Portugal. isapintopais@gmail.com
III Department of Pediatric Reumatology, Centro Hospitalar Vila Nova de Gaia/Espinho. 4400-129 Vila Nova de Gaia, Portugal. lucia.trindade.rodrigues@gmail.com
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
Juvenile dermatomyositis is an autoimmune systemic vasculopathy, mainly characterized by chronic inflammation of the striated muscle and skin. The authors report the case of a 15-year-old boy presenting with a four-month history of myalgia and proximal muscle weakness on the upper and lower limbs. These symptoms were associated with heliotrope palpebral exanthema, erythema in the dorsum of the hands, Gottron’s papules, erythematous and petechial rash on the extensor face of the thighs, and mild dysphagia for solids. Blood tests revealed an increase in muscle enzymes and electromyography showed changes suggestive of severe acute myopathy. Intravenous methylprednisolone was initiated, followed by a combination regimen of prednisolone and methotrexate. Progressive dysphagia, cutaneous abnormality, and muscular strength improvement were noted. With this case, the authors intend to raise awareness of a rare disease with an essentially clinical diagnosis, presenting in most cases with characteristic manifestations that should be recognised.
Keywords: exanthema; juvenile dermatomyositis; myositis
RESUMO
A dermatomiosite juvenil é uma vasculopatia sistémica de natureza autoimune, caracterizada por inflamação crónica do músculo estriado e da pele. É apresentado o caso de um adolescente do sexo masculino com um quadro de mialgia e fraqueza muscular proximal dos membros superiores e inferiores com quatro meses de evolução, associado a exantema palpebral em heliotropo, eritema purpúrico no dorso das mãos, pápulas de Gottron com atingimento peri-ungueal, exantema eritematoso nas coxas e disfagia ligeira para sólidos. O estudo analítico revelou aumento das enzimas musculares e a eletromiografia evidenciou alterações sugestivas de miopatia grave em fase aguda. O doente iniciou metilprednisolona endovenosa em bólus, seguida de um esquema de prednisolona e metotrexato, com melhoria progressiva da disfagia, força muscular e alterações cutâneas. Com este caso clínico, os autores pretendem alertar para uma doença rara com diagnóstico essencialmente clínico, que se apresenta na maioria dos casos com manifestações características que devem ser reconhecidas.
Palavras-chave: dermatomiosite juvenil; exantema; miosite
Introduction
Juvenile idiopathic inflammatory myopathies are a heterogeneous group of immune-mediated diseases. Juvenile dermatomyositis (JDM) is the most common clinical phenotype of myositis in children and adolescents, but remains a rare disease.1,2 Most studies suggest that JDM is an autoimmune angiopathy, resulting from exposure of genetically susceptible individuals to certain environmental triggers.3
JDM has an incidence of 0.55 cases per year and a prevalence of 6 in 100,000 inhabitants in Europe.2 Mean age of onset is 7.5 years and girls are mostly affected.1
Diagnostic criteria for JDM were established by Bohan and Peter in 1975 and include pathognomonic exanthema, proximal muscle weakness, raised muscle enzymes, myopathic changes on electromyography, and typical muscle biopsy.4,5 Current practice revealed the need of broadening classification criteria by incorporating new exams, such as magnetic resonance imaging (MRI), ultrasound, capillaroscopy, and myositis autoantibodies. These criteria are included in more comprehensive recommendations recently established by the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR).6,7
Corticosteroids, as prednisolone, are first-line agents in JDM treatment. However, recent studies suggest superior efficacy of a combined regimen of prednisolone plus methotrexate (MTX).1,7,8 Cyclosporine A, intravenous human immunoglobulin, cyclophosphamide, azathioprine, mycophenolate mofetil, hydroxychloroquine, tacrolimus, rituximab, infliximab, and autologous stem cell transplantation are possible alternatives used in the treatment of refractory disease.1,9-11
JDM prognosis is variable, with an overall mortality rate of 2−3%.1 Approximately 24−40% of patients experience a monocyclic course, recovering and successfully discontinuing medication within two years. Around 25% have a polyphasic course, with complete remission and recurrence periods. The remaining 50% of patients have chronic disease.1
Case report
A 15-year-old healthy boy was referred to the Pediatric Rheumatology Clinic due to progressive asthenia, myalgia, and muscle weakness on the upper and lower limbs, with proximal predominance, during the previous four months. Muscular symptoms worsened in the previous two weeks, with gait difficulty and muscle weakness in daily life activities. The patient additionally presented erythema of the dorsal surface of the hands together with malar rash, eyelid exanthema, and periorbital edema with one month of evolution. Two weeks later, he noticed a rash in the anterior surface of lower limbs. Importantly, the rash worsened with sun exposure. The patient also mentioned mild dysphagia for solids with a week of evolution. He denied fever, arthralgia, anorexia, weight loss, hematemesis or melena, abdominal pain, respiratory symptoms, or Raynaud’s syndrome.
On physical examination, the boy had good general condition, with stable vital signs. He presented palpebral exanthema in heliotrope associated with periorbital edema (Figure 1) and purpuric erythema in the dorsum of the hands, periungual telangiectasias and Gottron’s papules on metacarpophalangeal and interphalangeal joints (Figure 2), and erythematous and petechial rash on the extensor face of the thighs. Additionally, he presented symmetrical reduction of proximal muscle strength in upper and lower limbs and positive Gower’s signal, with Childhood Myositis Assessment Scale (CMAS) score of 28.
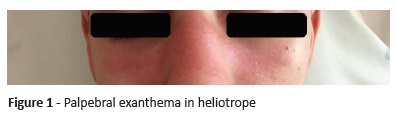
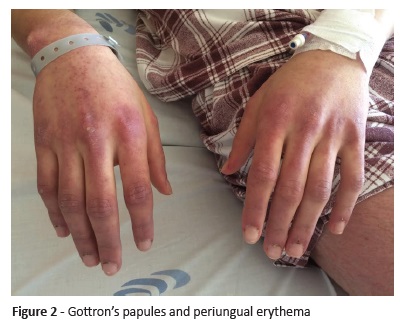
Laboratory tests revealed aspartate aminotransferase 267 U/L (Normal values 4-35), alanine aminotransferase 175 U/L (4-26), lactate dehydrogenase 777 U/L (135-225), creatine kinase 6,390 U/L (26-174), myoglobin 759 ng/mL (28-72), and aldolase 55.7 U/L (0-7.6). Blood count and sedimentation rate were normal, as were C-reactive protein and renal function. Serological tests (Cytomegalovirus, Rubeola, Toxoplasmosis, Herpes Simplex I and II, Mycoplasma pn, Chlamydia pn, Legionella pn, Parvovirus, Epstein-Barr virus, Borrelia burgdorferi, and Hepatitis B and C) were all negative.
Autoimmunity study revealed normal C3 and C4, 1/80 title of anti-nuclear antibodies (ANA) with mottled pattern. Anti-neutrophil cytoplasmic antibodies (ANCA), myositis-specific antibodies, anti-Mi-2, anti-SRP, anti-Jo-1, anti-PL-12, anti-PL-7, anti-OJ, anti-EJ, as well as myositis-associated antibodies, anti-Scl, anti-Ro-52, and anti-Ku were all negative.
Electromyography (EMG) revealed spontaneous denervation activity on examined muscles, with typical features of severe acute myopathy. Electrocardiogram, echocardiogram, chest radiograph, and functional respiratory tests were normal.
Capillaroscopy showed architectural disorganization, giant capillaries, capillary loss, and angiogenesis.
Juvenile dermatomyositis diagnosis was established and the patient was treated with intravenous methylprednisolone for five days (30 mg/kg/day) during hospitalization. Muscle strength improvement was observed, together with slight improvement of cutaneous manifestations and dysphagia. The patient was discharged with oral prednisolone 1 mg/kg/day, calcium, and vitamin D. Sun protection was recommended and physiotherapy was started. Oral methotrexate 12.5 mg/m2 (20 mg) was added two weeks after. One month after starting the combined regimen, almost complete muscle strength recovery (CMAS score of 48) as well as dysphagia and skin rash improvement were observed, with no evidence of calcinosis or other complications. Six months later, methotrexate was changed to subcutaneous administration for therapeutic optimization. Regimen was progressively reduced and after eight months of treatment symptoms completely resolved (CMAS score of 52).
The patient completed two years of combined treatment, without disease relapse. After three years of follow-up, he remains asymptomatic, without treatment, and is followed as an outpatient in the Pediatric Rheumatology Clinic.
Discussion
The authors presented a clinical case of JDM diagnosed at 15 years of age in a male patient. In JDM, symptom onset is more frequent at earlier ages and prevalence is higher in females.3 In this case, no putative triggering factors were identified.
Diagnostic criteria for JDM are essentially based on clinical manifestations, according to the EULAR/ACR Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies.6 This patient presented the characteristic heliotrope eyelid cutaneous rash associated with periorbital edema, as well as Gottron’s papules on metacarpophalangeal and interphalangeal joints. He also displayed proximal and symmetrical muscular weakness at shoulder and hip level, with difficulty in standing up, as confirmed by CMAS score.
Furthermore, complementary diagnostic tests revealed an increase in muscle enzymes and EMG changes suggestive of myopathy and denervation. Presently, EMG is not mandatory to establish diagnosis, and other exams, as total-body MRI, can be used to corroborate JDM diagnosis and help monitor disease activity. Muscle biopsy is also not mandatory to establish diagnosis, being mainly recommended when presentation is atypical or diagnosis is challenging, and was not performed in the present case.7 Capillaroscopy is increasingly used when severe capillary abnormalities, such as giant capillaries with intense neoangiogenesis, are common.12
Although the role of antibodies in JDM pathogenesis has not been clarified, its value is increasingly acknowledged due to association with specific phenotypes, complications, and prognosis. Around 70% of patients are estimated to present myositis-specific or myositis-associated antibodies.1,13 In this patient, all such autoantibodies were screened and negative. Regarding other autoantibodies analyzed, 1/80 titer for ANA should be highlighted, although ANA values are variable in JDM.14
Moderate-to-severe striated muscle damage, with limitation of daily activities and dysphagia prompt hospitalization for intravenous corticosteroid therapy.15 In the present case, combined prednisolone and oral MTX regimen was started early in treatment course, with muscle weakness improvement and cutaneous attainment partial improvement. Due to dysphagia persistence, MTX was changed to subcutaneous administration, with complete symptom resolution. This would have been the best option from the beginning, and the patient should have been initially treated with prednisolone and subcutaneous MTX.
Conclusion
This clinical case illustrates the challenging JDM diagnosis. Despite the condition’s rarity, JDM diagnosis remains essentially clinical, often presenting with typical clinical disease manifestations. It is therefore crucial to consider JDM in the differential diagnosis of pediatric myopathies and cutaneous rashes. Early recognition and treatment improve disease prognosis, preventing onset of calcinosis and other major complications.
REFERENCES
1. Rider GL, Katz JD, Jones OU. Developments in the Classification and Treatment of the Juvenile Idiopathic Inflammatory Myopathies. Rheum Dis Clin North Am. 2013; 39:877-904. [ Links ]
2. Prevalence and incidence of rare diseases: Bibliographic data. Orphanet Report Series. Rare Diseases Collection. January 2018. [ Links ]
3. Rider GL, Lindsley CB, Miller FW. Juvenile Dermatomyosites. Textbook of Pediatric Rheumatology. Chapter 26, p. 351-83. Elsevier, Seventh edition. [ Links ]
4. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975; 292:344-7. [ Links ]
5. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975; 292:403-7. [ Links ]
6. Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups. European League Against Rheumatism/American College of Rheumatology Arthritis Rheumatol. 2017; 69:2271-82. [ Links ]
7. Enders FB, BaderMeunier B, Baildam E, Constantin T, Dolezalova P, Feldman MF, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis 2017; 76:329-40. [ Links ]
8. Ruperto N, Pistorio A, Oliveira S, Zulian F, Cuttica R, Ravelli A, et al. Paediatric Rheumatology International Trials Organisation (PRINTO). Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet 2016; 13;387:671-8. [ Links ]
9. Inocencio J, Enríquez-Merayo E, Casado R, González-Granado LI. Subcutaneous Immunoglobulin in Refractory Juvenile Dermatomyositis. Pediatrics. 2016; 137. pii: e20153537. [ Links ]
10. Fasano S, Gordon P, Hajji R, Loyo E, Isenberg DA. Rituximab in the treatment of inflammatory myopathies: a review. Rheumatology (Oxford). 2016; 2.pii: kew146. [ Links ]
11. Moghadam-Kia, Oddis CV, Aggarwal R. Modern Therapies for Idiopathic Inflammatory Myopathies (IIMs): Role of Biologics. Clin Rev Allergy Immunol. 2017; 52:81-7. [ Links ]
12. Chojnowski MM, Felis-Giemza A, Olesińska M. Capillaroscopy - a role in modern rheumatology. Reumatologia 2016; 54:67-72. [ Links ]
13. Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford). 2009; 48:607-12. [ Links ]
14. Holden DJ, Brownell AKW, Fritzler MJ. Clinical and serological features of patients with polymyositis or dermatomyositis. Can Med Assoc J. 1985; 132:649-53. [ Links ]
15. Carbonell MS, Antoli HC, Fonseca AL, Talayero JM, Tomás PE. Dermatomyositis. Presentation of a mild to moderate case with early dysphagia. An Pediatr (Barc) 2015; 82:e86-9. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Mariana Capela
Department of Pediatrics
Centro Hospitalar Vila Nova de Gaia/Espinho
R. Dr. Francisco Sá Carneiro
4400-129 Vila Nova de Gaia
Email: marianacarvalhocapela@gmail.com
Received for publication: 29.04.2018. Accepted in revised form: 25.11.2019

