











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Congenital disorders of glycosylation (CDGs) are a highly variable, rapidly expanding family of genetic diseases resulting from defects in the synthesis of glycans that either lead to underglycosylation or misglycosylation of glycoconjugates.1 Although the vast majority of these monogenic diseases are inherited in an autosomal recessive way, about 15 CDGs show an autosomal dominant or X-linked inheritance. Some show either one or the other form of inheritance depending on the variant(s).2,3
The first patients with CDGs were reported in 1980 by Jaeken et al., being currently identified more than 140 forms of the disease. 2,4The exact prevalence and incidence of CDGs as a group have not been established. Estimates point to a prevalence of 1/10,000 in Europeans and African- Americans based on carrier frequencies of known pathogenic variants in 53 genes. However, most CDG types have less than 100 cases reported worldwide.2 Due to the difficulties in diagnosing this type of genetic diseases, their true prevalence is probably underestimated.
Oligosaccharides, or glycans, are multisugar structures attached to proteins or lipids. Glycosylation is an extremely variable and ubiquitous process that refers to the attachment of glycans to proteins and lipids to form glycoproteins and glycolipids. It is a crucial step in the processing of these enzymes, as it is required for folding, transport, self-recognition and function. Hypoglycosylation of glycoproteins acting as hormones, enzymes or transporters leads to its malfunctioning, decreased activity and rapid degradation.1,5 Given the universal nature of glycosylation in human biology, it is not surprising that disorders in this pathway can manifest as severe, usually multisystemic diseases.6
Objectives
The aim of this study was to review the current state of the art, namely regarding available therapeutic options of CDGs and present a simplified diagnostic approach to this group of disorders.
Etiology
Attachment of glycans to proteins can be of two types: N-linked glycosylation (attachment to the amide group of asparagine via an N-acetylglucosamine residue [GlcNAc]) or O-linked glycosylation (attachment to the hydroxyl group of serine or threonine via an N-acetylgalactosamine [GalNAc] or another monosaccharide residue). The synthesis of N-glycans requires a much longer and more complex pathway than that of O-glycans, which includes N-glycosylation and a processing pathway that is absent in O-glycosylation. N-glycosylation occurs in three cellular compartments: cytosol, endoplasmic reticulum (ER) and Golgi apparatus. Figure 1 depicts a schematic view of the initial steps of the N-glycosylation pathway.1,7,8
The biosynthesis of O-glycans usually starts in the Golgi apparatus with the attachment of GalNAc (or xylose in the case of glycosaminoglycans) to the hydroxyl group of serine or threonine residues.1 Contrary to N-glycan synthesis, no processing is involved.7
Lipid-based glycosylation pathways may also be affected in CDGs. The disorders that disturb the glycosylphosphatidylinositol (GPI) anchor biosynthesis pathway are a major group of glycosylation disorders that affect glycolipid production.8
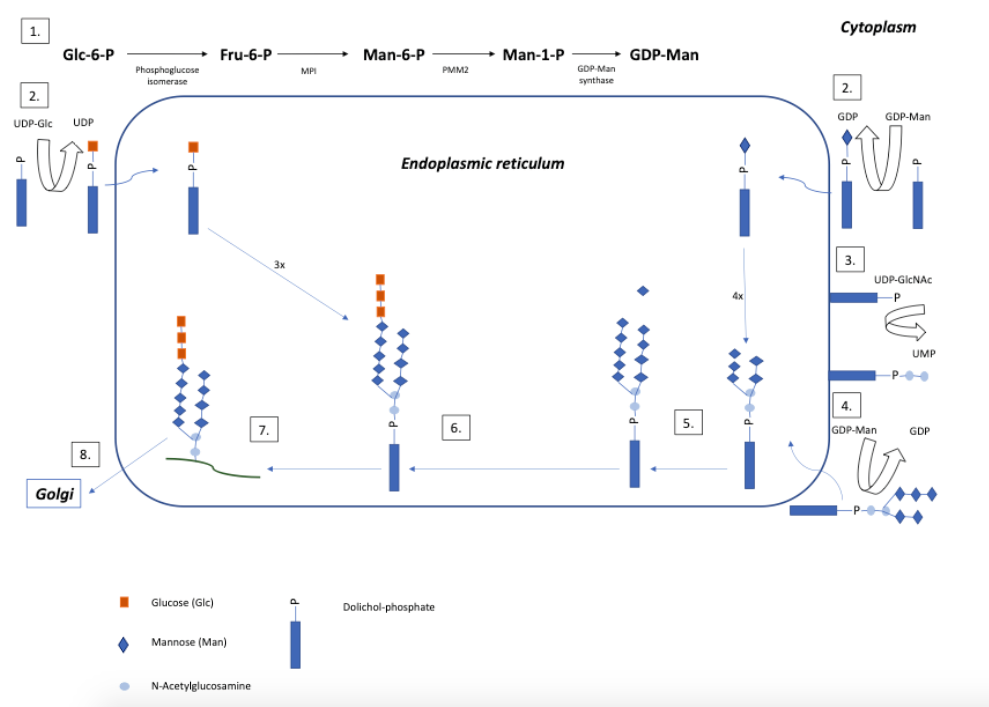
Figure 1 Simplified view of the initial steps of the N-glycosylation pathway (Adapted from Vilarinho L et al.1 and Grünewald S et al.4 - Frut: fructose, GalNAc: N-acetylgalactosamine, GDP: guanosine diphosphate, GDP-Man: guanosine diphosphate- mannose, Glc: glucose, GlcNAc: N-acetylglucosamine, Man: mannose, MPI: mannose phosphate isomerase, P: phosphate, PMM: phosphomannomutase, UDP: uridine diphosphate, UMP: uridine monophosphate
Physiopathology
Disorders can either be caused by abnormal assembly and transfer of oligosaccharides onto growing proteins, usually in the ER, or by abnormal processing of bound oligosaccharides in the Golgi apparatus.5 The multisystemic manifestations of CDGs can be explained by the fact that N-linked glycoproteins represent more than 10% of all proteins. The importance of O-glycosylation is best exemplified by the O-mannosylation of dystroglycan, which is required for normal muscular scaffolding formation, and by the O-xylosylation of glycosaminoglycans for proteoglycans, which plays a role in connective tissue.1,8,9
GPI anchoring is crucial for signal transduction, cell adhesion, and antigen presentation.8
The signs and symptoms associated with CDGs are either due to substrate accumulation, decrease of a final reaction product, or a combination of both. Glycosylation of proteins and lipids is a ubiquitous and extremely diversified process that modifies the intrinsic properties of these molecules, accounting for the different physiopathological mechanisms that give rise to the clinical manifestations of CDGs.1
Nomenclature
Historically, CDGs were named according to the patterns of transferrin isoelectric focusing (type I vs. type II) and alphanumerically according to their first description. Type I N-linked disorders were termed CDG-I “x” and type II disorders were termed CDG-II
“x”, with the “x” being alphabetically assigned according to the discovery order (e.g., CDG-Ia, CDG Ib).5 The N-linked protein glycosylation defect PMM2-CDG, previously known as CDG type Ia, was the first CDG to be described by Jaak Jaeken in 1980. With the widespread use of next-generation sequencing (NGS) in diagnosis, CDG nomenclature was updated in 2009 to specify the molecular etiology of the diseases. Presently, the disorders are denoted by the name of the affected gene (non-italicized), followed by the designation CDG (e.g., PMM2-CDG).2,5,10
Classification
CDGs are broadly classified into four categories: 11
(I) N-linked glycosylation defects
(II) O-linked glycosylation defects
(III) Combined glycosylation defects (multiple pathway involvement)
(IV) Glycosphingolipid and glycosylphosphatidylinisotol (GPI) anchor synthesis defects
Clinical manifestations
Due to the diversity of glycosylation pathways and targeted proteins, CDGs are usually multisystem diseases. Like in other inborn errors of metabolism, the phenotype may range from mild to severe, depending on the disease severity.8,12 Some clinical features may present at birth and remain unchanged or improve over the course of time, while others are progressive and may not be evident during infancy.8
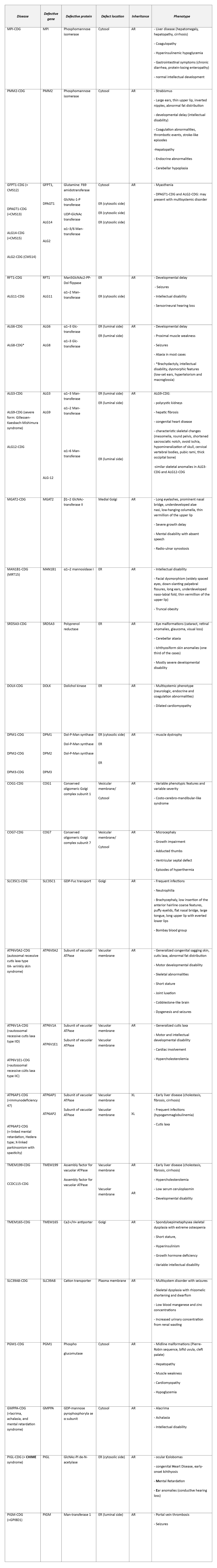
Table 1 presents a summary of selected CDG subtypes.13
Table 1 Overview of selected congenital disorders of glycosylation types of CDG. (Adapted from Ferreira CR et al.18)
Dysmorphic features
Although CDGs usually carry no complications during pregnancy or birth, the association with nonimmune hydrops fetalis has been reported.14 Newborns may present with facial dysmorphism (prominent cheeks, large and dysplastic ears), abnormal subcutaneous adipose tissue distribution (orange peel skin, fat pouches, inverted nipples), skeletal abnormalities, or dystrophic limbs. Children with PMM2-CDG typically present abnormal fat distribution and inverted nipples, but other forms of CDGs (ALG12-CDG, DPAGT1-CDG, MGAT2-CDG, MOGS-CDG) have also been associated with dysmorphic features. Dysmorphic facial features associated with PMM2-CDG have been described in large series and include prominent forehead, large ears and ear lobules, almond-shaped eyes, thin upper lip, prominent jaw, and long and slender fingers and toes.13 It is reasonable to consider the diagnosis of CDGs in patients with dysmorphic features and no clear diagnosis.1,5
Neurologic involvement
Due to the high biosynthetic demand of the central nervous system, neurologic manifestations are present in almost all CDGs.8 Deficient glycosylation in neural development may be relevant for the development and maintenance of normal cognitive functions.16 While some CDGs have been associated with a purely neurologic phenotype, others show neurologic involvement as part of a multisystemic presentation. MPI-CDG, one of the few treatable forms of CDGs, is a notable exception, as it does not present with neurologic involvement.5 The most common symptoms are psychomotor retardation/intellectual disability, hypotonia with frequent hyporeflexia, microcephaly, epileptic seizures, ataxia/cerebellar syndrome, peripheral neuropathy, spasticity, nystagmus, retinitis pigmentosa, strabismus, and stroke-like episodes.11,12,16 Apart from cerebellar and cerebral atrophy, most CDG patients with epilepsy have no characteristic brain malformations. Nevertheless, O-glycosylation disorders are associated with features such as lissencephaly, polymicrogyria, schizencephaly and neuronal heterotopia. The cerebellum is often affected in PMM2-CDG, dystroglycanopathies, and SRD5A3-CDG, although the course of cerebellar ataxia is not progressive.12,16 Cerebellar vermian hypoplasia is considered a highly specific sign of CDGs, but rarely present in the neonatal period.
Mutations affecting O-linked glycosylation result in congenital muscular dystrophy-dystroglycanopathies, including Walker-Warburg syndrome and muscle-eye-brain disease. This spectrum of diseases encompasses structural brain defects, such as lissencephaly, midline defects, cerebellar hypoplasia, hypotonia/myopathy and a spectrum of ocular and retinal conditions.5,8
GPI anchoring biogenesis defects can present as multiple congenital anomaly syndromes, with severe neurologic involvement and early death.8
Musculoskeletal involvement
Because collagen is a glycoprotein, CDGs can present with connective tissue or skeletal abnormalities. Several CDGs have skeletal dysplasia as the most prominent feature, including ALG12-CDG, TMEM165CDG and COG7-CDG.5 CDGs with well-defined skeletal dysplasia include ALG3-CDG, ALG6-CDG, ALG9-CDG, PGM3-CDG, CSGALNACT1-CDG and SLC35D1-CDG. Some skeletal abnormalities are also unique to particular CDG subtypes such as Schneckenbecken dysplasia in SLC35D1-CDG, brachytelephalangy in PIGV-CDG and PIGO-CDG, pseudodiastrophic dysplasia in ALG12-CDG, Gillessen-Kaesbach and Nishimura skeletal dysplasia in ALG9-CDG and Desbuquois dysplasia in PGM3-CDG.12 Ehlers-Danlos syndrome, progeroid type characterized by joint hypermobility, loose skin, short stature, osteopenia, and poor wound healing, is now classified as a CDG. Hereditary multiple osteochondroma syndromes, which are inherited in an autosomal dominant manner and can present with osteochondromas at birth in 5% of cases, are also included in the group of CDGs.5
Dermatologic involvement
Various skin abnormalities can be found in CDGs, including the characteristic “orange peel”, fat pads, ichthyosis, increased skin laxity, tumoral calcinosis, hypo/hyperpigmentation, aplasia cutis congenita and hypohidrosis/hyperthermia. Cutis laxa type IIA is now recognized as a CDG.5,15
Gastrointestinal and liver involvement
A common feature of many CDGs is failure to thrive, which has a probable multifactorial etiology comprising orofacial motor dysfunction secondary to hypotonia, malabsorption, neurologic impairment and alterations in the highly glycosylated gastrointestinal mucosa. Lymph circulation may also be abnormal in some patients. Reflux and vomiting are commonly found in CDG patients, and chronic diarrhea can also be found in several CDG types.11 Protein-losing enteropathy is a specific feature commonly found in MPI-CDG, but occasionally also present in other forms, such as ALG6-CDG and PMM2-CDG. Ascites is a critical CDG manifestation, with hypoalbuminemia being a potentially significant complication of these conditions, often linked to poor prognosis in severe infantile cases.5,11
The liver is a major site of glycosylation in the body, as it produces most glycosylated proteins. Therefore, glycosylation disorders are expected to affect not only liver development but also its structure and function. Liver involvement is associated with one of two main pathophysiological patterns: developmental abnormalities (ductal plate malformation or congenital hepatic fibrosis, as in MPI-CDG), or a combination of elevated liver transaminases (more common), hepatomegaly (less common), and steatosis in early infancy/childhood, as in PMM2-CDG. While the former is only treatable with liver transplantation, the second may spontaneously resolve over time. The most severe form of liver disease (acute liver failure) can arise in the setting of multiorgan failure (COG7-CDG, ALG3-CDG) or severe multisystemic disease. No typical histologic pattern of liver disease is described in CDGs. Liver fibrosis and cirrhosis have been reported in PMM2-CDG, MPI-CDG, and TMEM199-CDG setting.11,12,17
Liver involvement has been estimated in about 22% of CDG types and can be debilitating or even life-threatening with progression to liver cirrhosis or liver failure. CDGs with predominant or isolated liver involvement include MPI-CDG, CCDC115-CDG, TMEM199-CDG, and ATP6AP1, all of which include the V-type ATPase complex in Golgi apparatus and are classified as type II CDGs. Other CDGs associated with liver disease include six N-glycosylation diseases (including PMM2-CDG) and seven CDG types with defects in multiple glycosylation pathways.11,17
Cardiac involvement
Cardiovascular manifestations are not typical features in most CDGs. Nonetheless, congenital heart defects or anomalies (specifically conotruncal defects) have been described in PMM2-CDG, SRD5A3-CDG and COG1-CDG. Cardiomyopathy (hypertrophic and dilated) and pericardial effusion have been reported in association with several forms of CDGs, particularly PMM2-CDG, with the latter ranging from mild and asymptomatic to more severe and with worse prognosis.5
Hematologic involvement
Although primary and secondary dysfunction due to liver disease can be difficult to distinguish, the most serious cause of morbidity and mortality in CDGs is hematologic dysfunction.5 This dysfunction arises from abnormal glycosylation of coagulation factors and platelet membrane glycoproteins, with an increase in the risk of thrombotic and bleeding complications, especially in PMM2-CDG, MPI-CDG, and ALG1-CDG. An imbalance between procoagulant and anticoagulant factors and nonspecific or dysfunctional platelet interactions accounts for coagulation abnormalities in CDGs. Low levels of factors IX and XI, antithrombin, protein C, and protein S, deficiency of factors II, V, VII, VIII, and X, prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT) and elevated D-dimer are common findings. Hematologic complications include arterial and venous thrombosis, mucosal and visceral hemorrhage, and stroke-like episodes of so far unclear etiology.5,11 Still, coagulation parameters can improve and even normalize over time, lowering the risk of clotting and bleeding events in adults. While decreased antithrombin, protein C, and protein S levels are associated with increased risk of thrombosis, the correlation between PT, aPTT, and levels of factors IX and XI is less clear.11
Endocrine involvement
CDGs can affect multiple endocrine pathways, including those involved in growth, thyroid function, glucose metabolism, and sexual development. Poor nutrition and/or dysfunction of the growth hormone/insulin-like growth factor I cascade may lead to growth failure, which is seen in most CDGs. Low normal to increased serum levels of growth hormone (GH), with decreased levels of insulin-like growth factor (IGF-1), suggest that CDG patients are often GH-resistant. Increased clearance of hypoglycosylated forms of insulin-like growth factor binding protein 3 (IGFBP-3) results in low levels of this hormone.11 On the other hand, elevated thyroid-stimulating hormone levels are frequently found in young patients due to receptor glycosylation defects, especially in PMM2-CDG. Although thyroid-binding globulin and total T4 levels are often decreased in CDGs, patients are usually clinically euthyroid.5,11 As adrenocorticotropic hormone is glycosylated, hypoglycemia, often associated with hyperinsulinism and adrenal insufficiency, may occur and present with lethargy, vomiting, or seizures. Female CDG patients often have elevated follicle-stimulating hormone (FSH) and luteinizing hormone levels but low estradiol, resulting in abnormal pubertal development, amenorrhea, and hypergonadotropic hypogonadism. Male CDG patients may present with small testes, elevated FSH, and cryptorchidism, with pubertal abnormalities being generally less common in males.11
Immunologic involvement
Normal functioning of cell-surface receptors, antibodies, and other critical components of the innate and adaptive immune systems depend on glycosylation. Therefore, immunologic dysfunction has been reported either as a major feature in several types of CDGs (ALG12-CDG, MAGT1-CDG, MOGS-CDG, PGM3-CDG, ATP6AP1-CDG/ATP6AP2-CDG, SLC35C1-CDG) or as a variable feature in more severe multisystemic presentations (PMM2-CDG).5,11 Frequent or severe infections (bacterial, viral, fungal) and inadequate antibody response to vaccination and sinopulmonary,gastrointestinal and skin infections are common. Neutrophilia or neutropenia, lymphopenia, and low immunoglobulin (Ig)A and G, levels are the most common laboratory abnormalities. ALG12-CDG may cause lethal sepsis secondary to hypogammaglobulinemia and B-cell dysfunction in the neonatal period. SLC35C1-CDG, or leukocyte adhesion deficiency, type II courses with recurrent bacterial infections. MAGT1-CDG is caused by a deficiency in magnesium transporter 1 and results in a form of X-linked primary combined immune deficiency (previously described as XMEN disease- X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia).5,11
Diagnosis
The heterogeneity of CDGs makes their diagnosis a challenge. As there is no universal or pathognomonic sign or symptom and no sensitive diagnostic test for all CDGs, it is crucial to keep a high index of suspicion of these diseases. It is reasonable to suspect of CDGs in all patients with unexplained clinical story, especially those with unexplained neurologic or multisystem disease.6 In general, patients presenting with one of the following clinical and biochemical features should undergo screening for glycosylation abnormalities: 5,12
nonimmune hydrops fetalis
inverted nipples, abnormal fat distribution
connective tissue involvement (such as cutis laxa)
unexplained multisystemic phenotype, including neurologic manifestations
non-progressive cerebellar ataxia
severe epileptic encephalopathy
elevated serum transaminases (especially if associated with decreased antithrombin/protein C and S activity)
mixed coagulopathy and hypercoagulability
hyperinsulinemic hypoglycemia
liver steatosis/fibrosis/cirrhosis of unknown etiology
recurrent pericardial effusion
cardiomyopathy
skeletal dysplasia (particularly pseudodiastrophic dysplasia, Gillessen-Kaesbach-Nishimura skeletal dysplasia, Desbuquois dysplasia, brachytelephlanagy)
arthrogryposis with congenital myasthenia-like phenotype
immunodeficiency
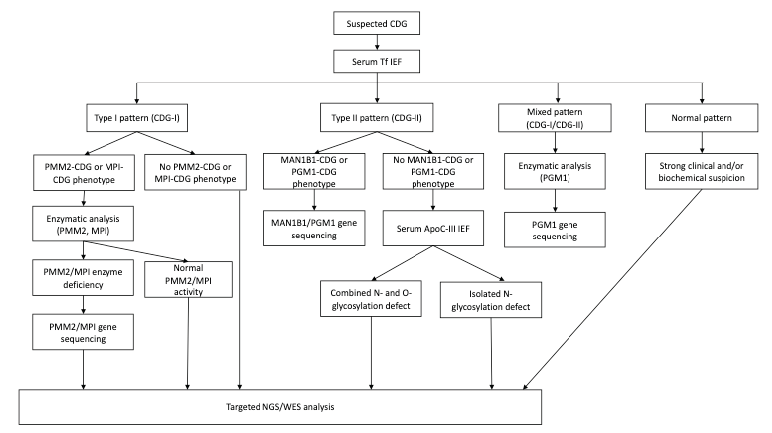
When a CDG is suspected, serum transferrin (Tf) isoelectric focusing (IEF) is the first step in screening.12 Transferrin is a plasma iron transport protein with two asparagine N-glycosylation sites. The dominant isoform in healthy individuals is tetrasialo-Tf, while asialo- and monosialo-Tf isoforms are usually not detectable. Failure in N-glycan synthesis causes partial sialic acid deficiency, changing the charge of serum transferrin and causing it to migrate towards the cathode in an electrophoretic field. A type I pattern (CDG-I) indicates an assembly or transfer defect of the dolichol-linked glycan in the cytosol or ER. A type II pattern (CDG-II) indicates a processing defect after glycan transfer in the ER or during glycosylation in the Golgi apparatus. Some CDGs, such as PGM1-CDG, display features of both serum Tf IEF patterns (mixed type, CDG-I/CDG-II).2,5,12,18
During CDG diagnosis, secondary causes potentially associated with abnormal glycosylation patterns should be excluded. False- positive results may occur with moderate alcohol consumption in a dose-dependent manner, Tf gene polymorphisms, end-stage liver disease, pregnancy, estrogen replacement therapy, classic galactosemia and fructosemia. False-negative results may occur at young ages (< 2-6 months), with abnormal glycosylation normalizing later in life. Furthermore, Tf IEF only detects isolated in N-glycosylation or combined N-and O-glycosylation defects.2,8,12
It should be kept in mind that a normal serum Tf IEF profile does not exclude CDGs. In cases of strong clinical and biochemical suspicion, targeted NGS or whole-exome sequencing (WES) should be considered since PMM2-CDG due to promotor defect and other CDGs (SLC35A1-CDG, SLC35A3-CDG, SEC23B-CDG, PGM3-CDG) may present with normal N-glycosylation patterns, and Tf IEF may be normal or altered in several other CDGs (as ALG13-CDG, SLC35A2-CDG, RTF1-CDG, and SRD5A3-CDG).12
The next step when evaluating a potential CDG is to determine whether Tf IEF shows a type I or type II pattern. If a type I pattern is present, phosphomannomutase-2 (PMM2) or phosphomannose isomerase (PMI) deficiency are the first hypotheses, and if the proper clinical setting is present, their enzymatic activity should be measured in fibroblasts or leukocytes. PMM2-CDG is the most common CDG and with the best-defined clinical phenotype, while MPI-CDG is treatable and potentially fatal if left untreated. If the clinical phenotype is not typical of PMM2-CDG or MPI-CDG and PMM2 and PMI activity is normal, NGS, including targeted NGS (panel of genes known to be associated with CDG), or WES should be performed. Biallelic pathogenic variants in PMM2 or PMI confirm the diagnosis of PMM2-CDG or MPI-CDG.2,12
A type II serum transferrin CDG pattern suggests the presence of a Golgi defect, with IEF of serum apolipoprotein C-III (apoC-III) recommended in these cases. Apo-CIII isoform analysis is a complementary test for type II CDGs, as it usually undergoes O-glycosylation with a mucin core 1 glycan. It is useful to distinguish between an isolated N-glycosylation defect and a combined disorder of N- and O-glycosylation.2,5,8,12
Matrix-assisted laser desorption/ionization (MALDI) coupled with time-of-flight mass spectrometry (TOF MS) allows to simultaneously assess the glycan structure of all glycoproteins in the blood (N- and O-linked glycans). This test is more sensitive for type II CDGs and also better able to distinguish between specific subtypes of CDG. However, glycosylation processes may be tissue-specific (especially in muscle and central nervous system), rendering this test less sensitive.2,5
Dystroglycan immunohistochemistry can be performed on muscle biopsy specimens. This test is particularly useful for the congenital muscular dystrophy spectrum, as the deficient chemical pathways in these diseases are only expressed in either brain or muscle tissue.5,8
Flow cytometry of blood granulocytes is used when lipid glycosylation and GPI anchor biosynthesis defects are suspected. Flow cytometry analysis of white or red blood cells for certain GPI-anchored cell surface proteins is available as a test for paroxysmal nocturnal hemoglobinuria (PNH) due to acquired mutations in the PIGA gene. The PNH test may also reveal abnormalities in other GPI anchor deficiencies.2
Molecular genetic testing is the most specific diagnostic test, particularly given that no biochemical test can screen for all CDGs. Molecular gene panel testing or exome sequencing may be performed even in the presence of normal screening results in cases of strong clinical suspicion. Biochemical and functional confirmation of molecular genetic findings is crucial since the number of new genes associated with CDGs is exponentially increasing, and most patients with CDGs carry at least one mild and often novel missense mutation.5,12
Figure 2 depicts a proposal of a diagnostic algorithm for CDGs.
Therapeutic approach
Specific treatment is only available for some types of CDGs, with symptomatic treatment being the only option for the majority. As most CDGs are multisystemic, a multidisciplinary approach is required to treat their several manifestations.12
Failure to thrive
The general approach consists of maximizing the tolerated caloric intake. Infants and children with CDGs can tolerate any type of formula, so no specific diet is required. Notwithstanding, elemental formulas may be better tolerated early in life. Feeding progression depends on the children´s oral motor function, with some requiring (transient) placement of a nasogastric tube or gastrostomy tube for nutritional support. However, failure to thrive often persists despite treatment.11,20
For patients with oral motor dysfunction and persistent vomiting and/or gastroesophageal reflux, postural measures, like maintenance of an upright position after eating and thickening of feeds, can be helpful. Speech and oral motor therapy support the transition to oral feeds and encourage speech when the child is ready.20
Protein-losing enteropathy
Regular albumin infusions, octreotide therapy, and diets rich in midchain fatty acids may be helpful in protein-losing enteropathy.11,12,20
Developmental delay
Case-by-case assessment should be performed to identify patients who may benefit from occupational therapy, physical therapy, or speech therapy.20
Ophthalmology
A consultation early in life with a pediatric ophthalmologist is important to identify potential eye abnormalities (strabismus, myopia, astigmatism, retinitis pigmentosa) and institute eye-preserving therapies (glasses, patching, or surgery), depending on the clinical situation.20
Hypothyroidism
Elevated thyroid-stimulating hormone (THS) levels are common, although CDG patients are usually euthyroid. Therefore, hypothyroidism should only be treated with levothyroxine in the presence of elevated TSH and low free thyroxine.11,12,20
Stroke-like episodes, coagulopathy and deep vein thrombosis
Supportive therapy for stroke-like episodes includes intravenous hydration, maintenance of normal blood glucose, and physical therapy during follow-up.20
Primary thrombosis prophylaxis is not recommended, but instead avoidance of potential triggers (elective surgery, oral contraceptives), adequate hydration, and early mobilization after invasive procedures.
Both unfractionated and low-molecular-weight heparin may be effectively used to treat thrombosis in the setting of CDGs. A reasonable alternative is factor Xa inhibitors. Vitamin K antagonists, such as warfarin, can be used for venous thrombosis secondary prophylaxis. 11
If necessary, fresh frozen plasma may be employed to correct factor deficiency and clinical bleeding.11,12,20
Immunologic status
In infants and children with a history of recurrent or severe infections, leukocyte count with differential and serum immunoglobulin levels should be retrieved at the time of diagnosis and during acute infection episodes. Unless otherwise indicated, age-appropriate vaccination is recommended.11,20
Orthopedic issues
Thorax shortening and scoliosis/kyphosis are examples of orthopedic issues experienced by some CDG patients. Orthopedic and physical medicine care, use of wheelchairs and transfer devices for home, and physical therapy are part of the management plan. Surgical treatment of these issues may occasionally be necessary.20
Specific treatment
Only a few types of CDGs have a specific treatment, usually consisting of supplementation with simple sugars with the aim of improving hypoglycosylation.
MPI-CDG is the most effectively treatable CDG. Treatment consists of mannose supplementation at the dose of 1 g/kg/day divided into four to six doses. Although treatment can significantly improve protein-losing enteropathy, liver disease may persist and progress. Caution is required during pregnancy.2,12,19
Although treatment of PMM2-CDG is mainly supportive, mannose-1-phosphate substrate replacement therapy is being investigated in clinical trials. 2,12 Acetazolamide has been recently shown to be effective in the treatment of cerebellar motor syndrome and improve coagulation parameters, clinical severity, ataxia, epilepsy and lipodystrophy in patients with PMM2-CDG.3,19 Some patients with SLC35C1-CDG respond to oral fucose supplementation, showing decreased incidence of recurrent infections with hyperleukocytosis but no neurodevelopment changes. D-galactose at a dose of 1.5-2.5 g/kg/day (until a maximum of 50 g/kg/day) has been shown to improve hypoglycemia, coagulopathy, and endocrinopathy in PGM-1-CDG. Galactose has also been suggested to improve endocrinopathy and coagulopathy in TMEM165-CDG and SLC39A8-CDG. Some patients respond to 15-20 mg/kg/day of manganese sulfate.2,12
Extended-release sialic acid and N-acetylmannosamine (ManCAc) are currently being investigated in GNE-CDG. Sodium butyrate has been shown to improve seizures in CAD-CDG and PIGM-CDG, and ketogenic diet has been shown to decrease seizure frequency in some PIGA-CDG patients.2,11,12,19 Uridine supplementation led to cessation of seizures and improvement in development and anemia in CAD-CDG.11,12,19
Organ transplantation may be an option for CDGs in which the primary manifestation is limited to a specific organ and progressive neurologic disease is not a concern.5. Bone marrow transplantation and hematopoietic stem cell transplantation are therapeutic options in PGM3-CDG.3
Advances over the last few years have opened new therapeutic avenues in CDGs. The most promising new approaches include activated sugars, pharmacological chaperones, and gene therapy.11
The delivery of mannose-1-phosphate poses a challenge as the molecule is highly unstable. Liposomal targeting could be an option for efficient liver targeting, but technical difficulties limit this approach. Using a large complex molecule as a capsule around the mannose-1-phosphate molecule could be an alternative, but its large size can potentially compromise an efficient cellular uptake. So far, none of these compounds have entered clinical trials.11
Proteins are present in the intracellular medium in folded or unfolded states. Point mutations may lead to a predominance of unfolded states, which lead to protein destabilization and aggregation. Pharmacological chaperones are small molecules that bind to specific proteins, stabilizing proteins in the folded state. PMM2-CDG has been classified as a misfolding disorder. In vitro testing has disclosed the possibility of uncovering ligands capable of stabilizing PMM2, suggesting that pharmacological chaperones may be a promising therapeutic option.11,21 Gene therapy consists of the transfer and activation of a fully functional copy of an aberrant gene to the patient´s system. Being monogenic diseases, CDGs are potential candidates for this approach.11,21 Apart from viral transgene delivery, such as adeno-associated virus vectors, other options include zinc-finger nucleases and transcription activator-like effector nucleases. Antisense therapy intends to restore transcript splicing after disruption by pathogenic splice variants. In vitro experiments have been conducted in PMM2-CDG patient-derived cells, and antisense therapy has shown promising results in TMEM165-CDG.11
Monitoring
The following laboratory tests and clinical assessments are recommended at the time of diagnosis (especially for PMM2-CDG) and subsequently on an annual basis:20
liver function and serum albumin tests
thyroid function tests (for TSH and elevated free thyroxine)
protein C, protein S, antithrombin III, factor IX
urinalysis
serum gonadotropins in adolescent and adult women, to assess hypogonadotropic hypogonadism
echocardiogram
renal ultrasound to evaluate bilateral hyperechogenic kidneys, enlarged kidneys and renal cysts (PMM2-CDG)
ophthalmologic examination
orthopedic evaluation when scoliosis becomes evident
Of note, acetaminophen and other liver-metabolized drugs should be carefully used in patients with liver involvement.20
Genetic counseling
Assessment of genetic risk and carrier status and discussion of prenatal testing are recommended before pregnancy. In cases of known pathogenic variant(s), prenatal diagnosis and preimplantation genetic testing for N-linked or multiple-pathway CDGs are possible.20
Assessment of relatives
It is acceptable to evaluate apparently asymptomatic older and younger siblings of CDG patients to establish a potential CDG diagnosis as early as possible. Assessment may include molecular genetic testing if the pathogenic variant in the family is known or serum transferrin analysis if the pathogenic variant is unknown and transferrin is abnormal in the affected patient.20
Prognosis
Despite multidisciplinary support, mortality from multiorgan failure or severe infections is reported in around 25% of CDG patients in the first years of life. Morbidity in these patients includes infections, seizures and hypoalbuminemia. Although there is no predictable life expectancy, the oldest living PMM2-CDG patients are currently in their 40s and 50s.2,22
Early CDG diagnosis is desirable since identification of the pathogenic variant(s) enables prenatal diagnosis and preimplantation genetic diagnosis of congenital disorders of N-linked glycosylation or multiple pathways.20
Take-home messages
CDGs are a vast group of heterogeneous diseases associated with elevated morbidity and mortality, especially in the first year of life. As defects may affect any organ at any age and elicit variable clinical presentations, their diagnosis poses a challenge.
CDGs should be part of the differential diagnosis in patients with multiorgan involvement, especially in the presence of neurologic or hepatic disease or congenital anomalies.
As there are no universally acknowledged signs or symptoms of CDGs, a high index of suspicion is crucial for diagnosis.

