











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Neurofibromatosis type 1 (NF1) is transmitted by autosomal dominant loss-of-function mutations in the neurofibromin gene of chromosome 17q11.1,2 Penetrance is almost 100%, with highly variable expression, even within the same family. It is one of the most common genetic disorders, with a reported incidence of 1:3000. Approximately one-half of cases are familial (inherited), while the remaining result from de novo (sporadic) mutations.1,2
The disease is multisystemic, and typical clinical manifestations comprise café-au-lait macules (CALM), axillary and/or inguinal freckling, Lisch nodules (iris hamartomas), choroidal abnormalities, neurofibromas, distinctive osseous lesions (sphenoid dysplasia, anterolateral bowing of the tibia, pseudarthrosis of a long bone), and optic pathway gliomas (OPG). The first NF1 diagnostic criteria reported date back to 1988 and did not allow to differentiate NF1 from conditions with overlapping phenotypes and distinct natural history, such as Legius syndrome and segmental/mosaic NF1. Since then, NF1 genetic testing has become clinically available, with high detection rates and clinically useful genotype-phenotype correlations.1 For this reason, diagnostic criteria have been recently revised and published (Table 1).1
Table 1 Revised diagnostic criteria for neurofibromatosis type 1 ─ International consensus recommendation
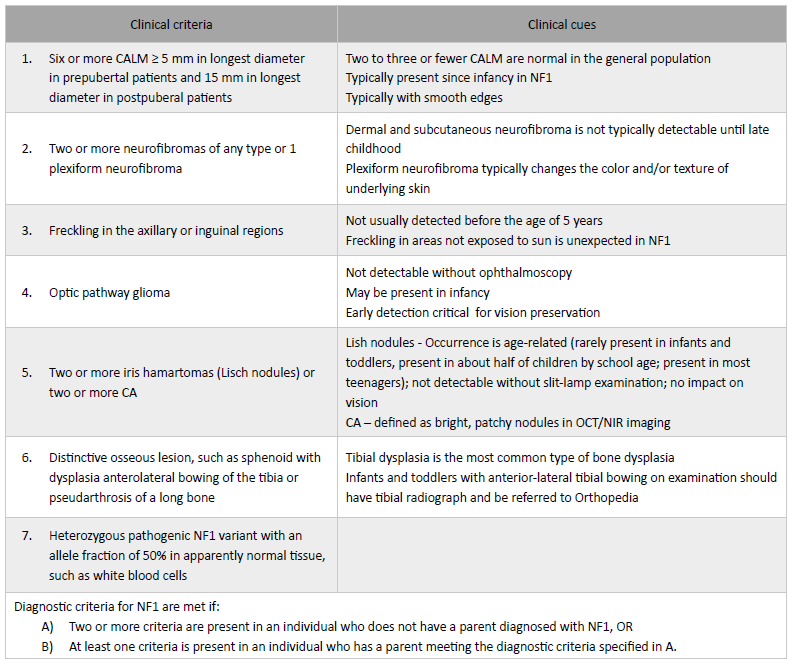
CA, choroidal abnormality; CALM, café-au-lait macule; NF1, neurofibromatosis type 1; NIR, near-infrared reflectance; OCT, optical coherence tomography. Adapted from Millus D et al(3) and Legius E et al 2021(1)
When a child fulfills the diagnostic criteria for NF1, the diagnosis is straightforward, and molecular genetic confirmation is usually not necessary.3 However, the diagnosis can be challenging, as clinical features evolve slowly and progressively and may often be absent in early childhood, and about 50% of NF1 patients have no family history.2,4 Therefore, NF1 genetic testing may enable higher detection rates by allowing to confirm a suspected diagnosis before the clinical diagnosis is feasible and distinguishing NF1 from conditions with similar phenotypes.4
Affected individuals are at risk of developing benign and malignant tumors, mainly of neuroectodermal origin (four times more often than in the general population; cit in Giovannoni I et al).5 OPG are the most common central nervous system-associated tumors, occurring in 15-20% of patients within the first decade of life.6,7 In NF1 children, OPG has a more benign course, with a largely indolent behavior, and may even regress without treatment.8 Despite the high survival rate, children with OPG still face important endocrine and visual sequelae.8
Endocrine disorders have been reported in 1-5% of all NF1 patients, specifically short stature, central precocious puberty (CPP), growth hormone (GH) and other pituitary hormone deficiencies and, rarely, GH excess, as well as obesity with insulin resistance/impaired glucose tolerance.9 Although endocrine disorders have been reported in these children at least since the seventies, sometimes leading to NF1 diagnosis, they are currently not included in the diagnostic criteria for the disease.1,4
Besides performing a periodic clinical checkup for detection of evolving known NF1 manifestations, as neurofibromas and skeletal abnormalities, pediatricians who follow NF1 children should also be aware of possible associated endocrinopathies.10
The aim of this article was to review the most frequent endocrine disorders in NF1 patients, what to monitor, and when to refer patients to pediatric Endocrinology consultation.
Pathophysiology of endocrine disorders
Loss of neurofibromin protein function leads to hyperactivation of the proto-oncogene RAS, as well as enhanced activity of RAS downstream effectors. RAS signaling pathway controls cell proliferation. Neurofibromin also positively regulates the generation of intracellular cyclic adenosine monophosphate (cAMP) in the brain. cAMP and the transcriptor factor known as cAMP response element-binding protein (CREB) are key regulators of hypothalamic-pituitary axis development.11
In the absence of organic or medical causes, NF1-associated pituitary hormone deficiencies may be explained by the loss of normal neurofibromin function and subsequent cAMP signaling impairment.12
Nonetheless, OPG is the most frequent underlying cause of endocrinological disturbances.11 In a study by Sani et al., at least one endocrinopathy was found in 55.6% of children with NF1 and OPG during a mean follow-up of 9.1 ± 3.4 years.8 None of the patients with OPG limited to the optic nerve developed endocrinopathy during follow-up, while 57.1% and 72.7% of patients with OPG involving the optic chiasm and hypothalamus developed endocrinopathy, respectively.
In the study by Santoro et al., OPG was diagnosed earlier in 26.7% of children with endocrine disorders compared to those without endocrine dysfunctions.6
Auxological features, growth, and puberty in NF1 children
Short stature and macrocephaly have long been acknowledged as clinical features of children with NF1.
Disease-specific growth charts for NF1 have been developed by Clementi et al. in 1999 (Figures 1, 2, 3 e 4).13 These charts are useful to determine the main growth profile differences in these patients compared to the healthy population, understand the pathogenesis of the condition, and allow the detection of a secondary growth disorder if present.13
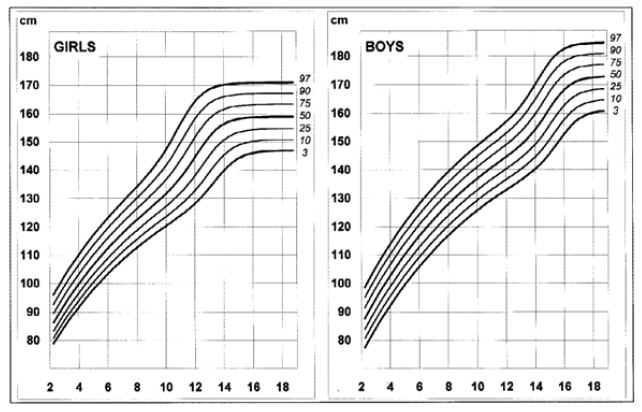
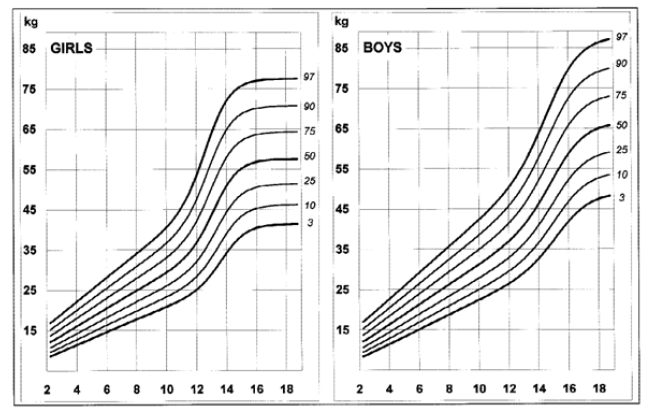
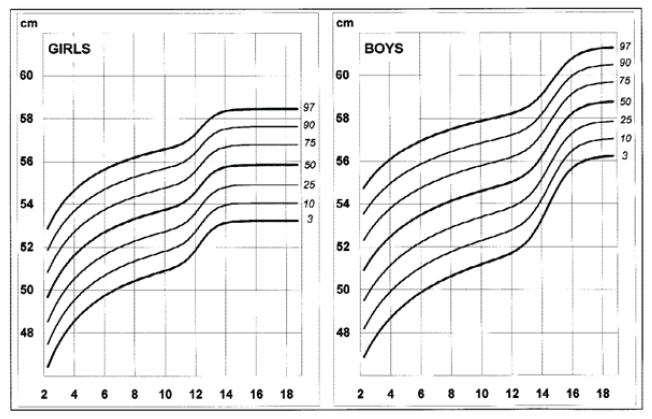
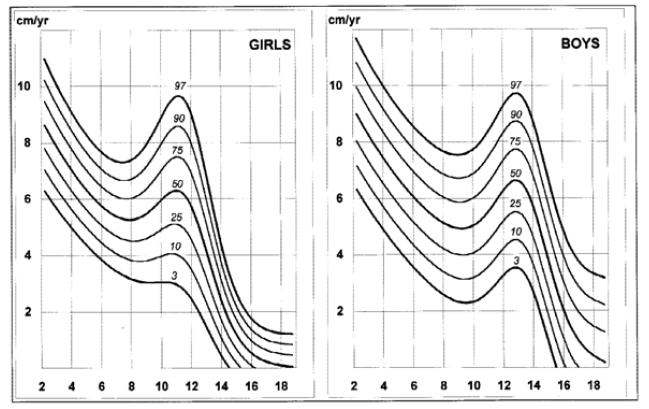
The growth profile of these children seems similar to their peers up to the age of seven years in girls and 12 in boys. Subsequently, the 50th centile tends to overlap with the 25th centile of healthy children, and the 3rd centile is significantly lower in NF1 subjects.13 Pubertal spurt occurs one year earlier compared to healthy controls and is mildly reduced in boys but not in girls. The study by Carmi D et al. showed the same pattern: a poor pubertal growth velocity (GV) and peak height velocity, particularly in boys, resulting in a significant and gradual reduction in the relative height for age during puberty.10 Most patients did not attain their expected height and ended up with a shorter standard deviation score (SDS) compared to their initial height SDS.
Short stature (height below -2 SDS) occurs in 13-24% of prepubertal children and in more than 40% of adults.10,14 Its etiology is probably multifactorial. It can be due to abnormal skeletal development, use of psychostimulant medications, short familial stature, hypothyroidism, precocious puberty, or GH deficiency (see below).
Head circumference (HC) is larger in NF1 patients during the whole growth period and in adulthood.13 The disproportion between head size and height increases with age, mainly in boys.13 A retrospective study in 80 Finnish NF1 children investigated the incidence and diagnostic accuracy of elevated HC to height ratio (HCHR). At a median age of NF1 diagnosis of 3.6 years, 53.8% of children showed elevated HCHR. Considering the seven diagnostic criteria for NF1, elevated HCHR was the second most common clinical manifestation after CALM (cit in Bizzarri C et al).11 Macrocephaly typically requires no particular follow-up.3
A disproportionate growth of the extremities, including leg-length discrepancy, is generally associated with plexiform neurofibromas, causing local bone growth acceleration and soft-tissue hypertrophy.3
Endocrine disorders in NF1 children
Central precocious puberty
Central precocious puberty (CPP) is the most common endocrine manifestation of NF1 in childhood.9,11 Its prevalence is higher than in the general population (~3-6% vs. 0.5%) and mainly affects boys.09,10,15 The condition is suspected due to the occurrence of breast budding (Tanner stage II) before the age of eight years in girls and testicular volume of at least 4 mL before the age of nine years in boys and is typically accompanied by GV acceleration.
CPP has been mostly reported in NF1 children with OPG, but it is not always so.9,15 In children without OPG, the occurrence of CPP may be incidental and idiopathic or result from NF1 itself, loss of normal neurofibromin function and impaired cAMP signaling, or mild cerebral abnormalities undetectable in magnetic resonance imaging, such as slow-growing hamartomas.11,16
In the study by Sani et al., around 20% of children with NF1 and OPG had both CPP and GHD.8 The coexistence of CPP and GHD can mask linear growth acceleration, resulting in normal GV and making GHD diagnosis more challenging. On the contrary, if GV remains high with appropriate CPP treatment, excess of GH should be suspected and investigated (see below). Since most children with OPG are asymptomatic, NF1 children should be closely monitored for early puberty signs leading to OPG diagnosis.15
Gonadotropin-releasing hormone (GnRH) agonist therapy should be decided on a case-by-case basis, as some patients do not benefit from this treatment. Long-term follow-up is required, since patients with OPG and precocious puberty can progress to gonadotropin deficiency later in life.8,14,17
Growth hormone deficiency
As previously mentioned, not only puberty but also growth is commonly affected in NF1 children, and disease-specific growth charts have been published. Although the associated mechanism is not clearly elucidated, growth hormone deficiency (GHD) is more common in patients with NF1 than in the general population. As with CPP, GHD is more frequent in the presence of intracranial tumors.11 In some cases, GHD is correlated with the use of radiotherapy, surgery, or chemotherapy as cancer treatments.6,9,11
The incidence of GHD is estimated to be 2.5%, with a mean age at diagnosis of nine years, but can be as high as 35% in NF1 children with OPG.8,9
A clear decrease in GV should raise suspicion of GHD, as well when growth is below the 3rd centile (particularly if perceived in disease-specific NF1 growth charts) and when the predicted adult height falls below the estimated mid parental height (mother’s height + father’s height + 13 cm in boys/2; mother’s height + father’s height - 13 cm in girls/2). In these cases, further research should be carried to rule out GHD.
Concerns have been raised about whether treatment with somatotropin can enhance tumor growth and/or increase malignancy risk, but no randomized clinical trials addressing these issues have been conducted so far.
GH receptor expression has been documented in plexiform neurofibromas. This finding could indirectly suggest that these tumors may respond to GH stimulation (Cunha et al., cit in Kyritsi EM et al.).12 Nonetheless, no increase has been noted in plexiform neurofibroma growth rate during puberty, a period of spontaneous increase in GH secretion (Dagalakis et al., cit in Kyritsi EM et al.).12
Howell et al. investigated a cohort of 102 NF1 patients receiving somatotropin treatment and reported a reassuring safety profile.18 Most adverse events reported seemed related to progression of NF1 features. CALM and neurofibroma changes were reported in 13% of patients over a median follow-up of 2.7 years, a percentage expected for disease progression in NF1 children.18 In the study by Sani et al., although tumor progression was not affected by somatotropin treatment, scoliosis progression could occur, which should be taken into account when planning treatment.8 Treatment response, although favorable, was not as good as in patients with idiopathic GHD.18
According to the current evidence, it seems very unlikely that the use of somatotropin affects the course of the several NF1 manifestations, in particular the risk of tumor development. GH treatment in NF1 patients with GHD seems beneficial, as it is associated with improved growth rate and is well tolerated.8,18
Published guidelines underline that somatotropin treatment should be reserved for children with poor GV and low GH stimulation documented through testing.3 According to the National Commission for the Normalization of Growth Hormone in Portugal, only one stimulation test with a peak of less than 7 g/L is required to initiate somatotropin treatment in children with short stature and auxologic criteria for somatotropin deficiency with an identified etiology (for example, OPG; unpublished data). In cases of idiopathic GHD, two stimulation tests are necessary.
Growth hormone excess
Somatotropin excess has also been described in NF1 patients. To date, there are no reports of somatotropin excess without imagiological evidence of OPG.7,11,19 The estimated incidence of somatotropin excess in children with NF1-OPG is 5-11%.6-8
Growth hormone excess is usually suspected by excessive/accelerated linear growth, with high levels of insulin growth factor 1 (IGF-1) and IgF-binding protein 3 and lack of GH suppression to levels <1 ng/mL after oral glucose tolerance test (OGTT) confirming the diagnosis. IGF-1 concentrations must be compared with age- and sex-dependent normative data.14 As there are no normative data in children, the diagnostic threshold of GH levels suppressibility during OGTT, especially during puberty, has not been clearly established.14 In the study by Cambasio et al., all patients studied for GHE and OPG had IGF-1 and IGFBP-3 levels above the 97th percentile. 7 Lack of GH suppression on OGTT was demonstrated in five of seven patients.
The mechanism underlying GH excess is still unclear. Infiltration of the somatostatinergic pathway by the tumor, presumably resulting in reduced somatostatin tone and overproduction of GHRH-mediated pituitary GH, has been proposed by Manski et al. (cit in Cambiaso P et al).7,14
In the study by Cambasio P et al., increased GV was the only sign of OPG in one patient, showing that it should be considered in the differential diagnosis of OPG and reinforcing the need for frequent and accurate auxologic evaluation in NF1 children.7 Because CPP is more frequent in this patient population, it is usually considered first in the differential diagnosis, often neglecting GH excess.19 However, CPP and GH excess may coexist. Therefore, when puberty is biochemically and clinically suppressed with GnRH agonist therapy and persistently high levels of IGF-1 are observed, particularly if combined with tall stature and growth acceleration, additional studies should be conducted to rule out GH excess.14
Concerns have been raised that endogenous GH excess may increase the risk of cancer in NF1 children and induce preexisting tumor growth, namely OPG.14,19Josefson et al. suggested that medical treatment of GH excess with somatostatin analogs (SSa) or GH receptor antagonists should be provided to reduce tumor growth and long-term detrimental systemic effects related to uncontrolled GH excess (gigantism-related skeletal problems, hypertension, glucose intolerance, and cardiomegaly).19
GH excess may be a transient phenomenon that resolves without treatment.7,14,19 In these cases, only short-term therapy with SSa may be required to reduce patient discomfort, treatment costs, and potential treatment side effects.
Pheochromocytoma
Pheochromocytoma (PHEO) is usually reported in NF1 adult patients with an estimated incidence of 0.1-5.7%. Its incidence in the pediatric population is unknown, but some cases have been reported in the literature.5 Classical symptoms include hypertension, headache, and hyperhidrosis (the three H’), with loss of circadian blood pressure rhythm characterized by a ‘non-dipping pattern’ as an early marker of future hypertension. Because not all NF1 patients with PHEO have hypertension, the lack of this sign should not be considered reassuring or taken as absence of the condition.11
Most experts recommend screening for PHEO if there is an acute and dramatic increase in heart rate and/or blood pressure.3
Gynecomastia
An increased frequency of unilateral and bilateral prepubertal gynecomastia cases has been described in NF1 patients, with endocrine workup shown to be normal in all.11 The management of gynecomastia in these patients should thus follow the same recommendations as for the general pediatric population.
Osteopenia and vitamin D deficiency
Childhood osteopenia is frequent in NF1, but the risk of fractures is only slightly increased. The etiology of osteopenia is unknown. It has been hypothesized that it may be related to decreased musculoskeletal strength and poor neuromuscular coordination, leading to decreased bone remodeling. 3 Children with NF1 have a high prevalence of insufficient serum (25-hydroxycholecalciferol) vitamin D levels.3 Periodic assessment of vitamin D levels, especially in the presence of risk factors (low sun exposure, use of antiepileptic drugs), is helpful in determining the need and dosing of vitamin D supplementation.
Discussion/conclusion
NF1 is a common genetic disorder with a wide spectrum of health implications. The first reports of endocrinopathies in children with NF1 date back to the 70s. At that time, endocrine disorders sometimes led to the diagnosis of NF1.4 Since 1988, when the first diagnostic criteria were published, the clinical diagnosis of NF1 has become increasingly familiar to pediatricians, who became vigilant for manifestations of this phacomatosis, such as CALM, freckling, neurofibromas, and others.20 Nowadays, NF1 diagnosis is performed earlier, often with the support of genetic testing, and endocrinological manifestations may sometimes arise during these children’s follow-up. CPP and GHD are the most common endocrine complications and often arise in the presence of seller and suprasellar tumors, such as OPG, representing their initial presentation.
General pediatricians and pediatricians of other subspecialties involved in the follow-up and management of these children should conduct a careful auxological assessment, as well as early assessment of pubertal signs and growth rate surveillance. Although NF1 children are, on average, shorter than children of the same age range, the use of specific growth charts, GV assessment, and mid parental estimation are crucial to identify secondary deviations that may benefit from referral and specific treatment.10,13 The endocrinological and treatment approach for these children do not differ from those of the general population.
In addition to periodic medical checkups for detection of evolving acknowledged NF1 manifestations (as neurofibromas and skeletal abnormalities), regular growth and puberty monitoring is also recommended in all patients (Table 2).3
Table 2 Health supervision guidelines for children with NF1
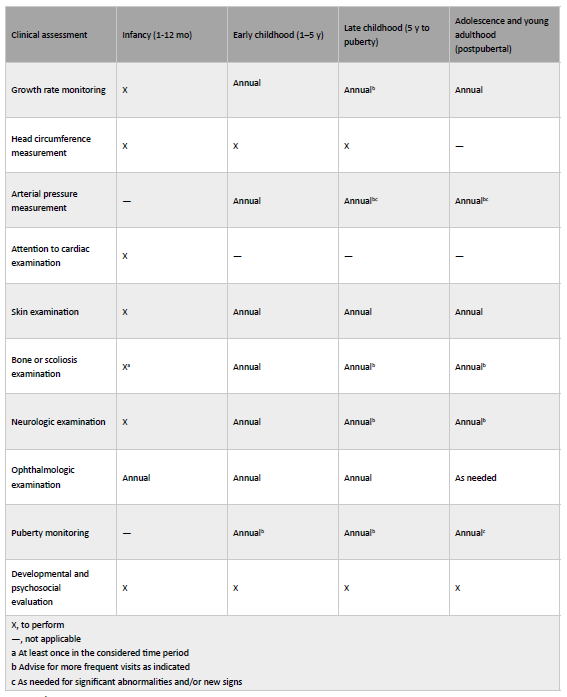
(adapted from Milles et al.(3)). Mo, month; y, year
When to refer children with NF1 to pediatric endocrinological consultation:
Breast budding before the age of eight years in girls and testicular volume of at least 4 mL before the age of nine years in boys.
Significant decrease in GV, growth below the 3rd centile or two SDS (particularly if perceived in NF1-specific growth charts) or when the predicted adult height falls below the estimated mid parental height.
Excessive/accelerated linear growth.
Cases of gynecomastia with one of the following features: larger than 4 cm; with fast progression; associated with rapid and early virilization; persisting beyond the age of 16 years.
Signs of insulin resistance, as acanthosis nigricans.
Obesity with comorbidities.
Detectable increase of heart rate and/or blood pressure.
Authorship
Maria Adriana Rangel - Conceptualization; Resources; Writing - original draft; Writing - review & editing;
Marta Vila Real - Resources; Writing - review & editing
Fátima Santos - Writing - review & editing
Ana Luísa Leite - Writing - review & editing
Rosa Arménia Campos - Writing - review & editing

