Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Cri-du-chat syndrome results from a deletion of variable size on the short arm of chromosome 5 (5p-).1,2) It is a rare entity that should be suspected in the presence of specific clinical features at birth, namely cat-like cry, low weight, microcephaly, and facial dysmorphism.2-5) It is also associated with delayed psychomotor development and presence of cardiac, neurological, or renal malformations, which should be investigated in routine workup.2,3) Herein is described the case of an infant who presented with cat-like cry since birth and whose genetic study was very challenging.
Case presentation
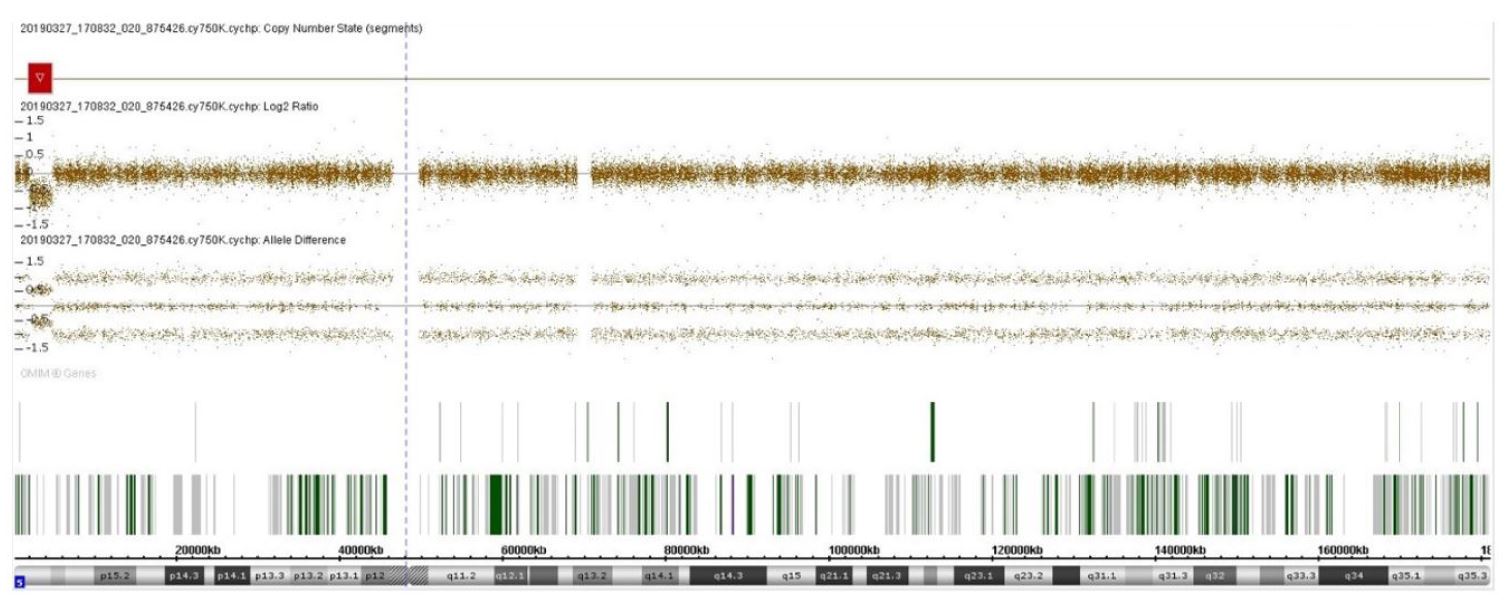
An eight-month-old female infant presented from birth with a high-pitched, cat-like cry and a peculiar face with wide nasal bridge and thin upper lip. She was born by eutocic delivery from a singleton pregnancy without complications and had an Apgar index of 9/10. Because of suspicion of Cri-du-chat syndrome, a genetic study was performed, first by fluorescence in situ hybridization (FISH) with a specific probe for the critical region of this syndrome at 5p15.2 (metasystem XL 5q31/5q33/5p15 FISH probe), which was negative for deletion of the short arm of chromosome 5. Following this result, chromosomal microarray analysis (aCGH) was performed, which revealed a deletion of 2.889 Mbp at 5p15.33p15.32 (genomic coordinates GRCh37: 1708529 to 4597389; Figure 1) involving MIR4277, MEPL36, NDUFS6, LOC101929034, IRX4, CTD-2194D22.4, LOC100506858, IRX2, C5orf38, LOC105374620, LINC01377, LINC01019, LINC01017 and IRX1 genes, suggesting a possible etiology for the observed phenotype. The patient was observed by a cardiologist, who excluded cardiac malformations. No morphologic changes were found in transfontanellar, abdominal, or renal ultrasound. At the time of the study, the infant was eight months old and presented with appropriate height, weight, and psychomotor development.
Discussion
Cri-du-chat syndrome results from a chromosomal abnormality with an estimated incidence of 1:15000 to 1:50000 live births.1-5 The condition has a mortality rate of approximately 10%, mainly in the first year of life.3 The classic phenotype is characterized by the presence of a high-pitched, cat-like cry early in life (apparently related to vocal cord abnormalities), facial dysmorphism (round face, wide nasal bridge, epicanthal folds), and microcephaly, as well as psychomotor and intellectual delay. Although a genotype-phenotype correlation has not been fully established, the disease has a variable spectrum of clinical features and severity that appears to depend on the size and gene content of the deletion.2-5
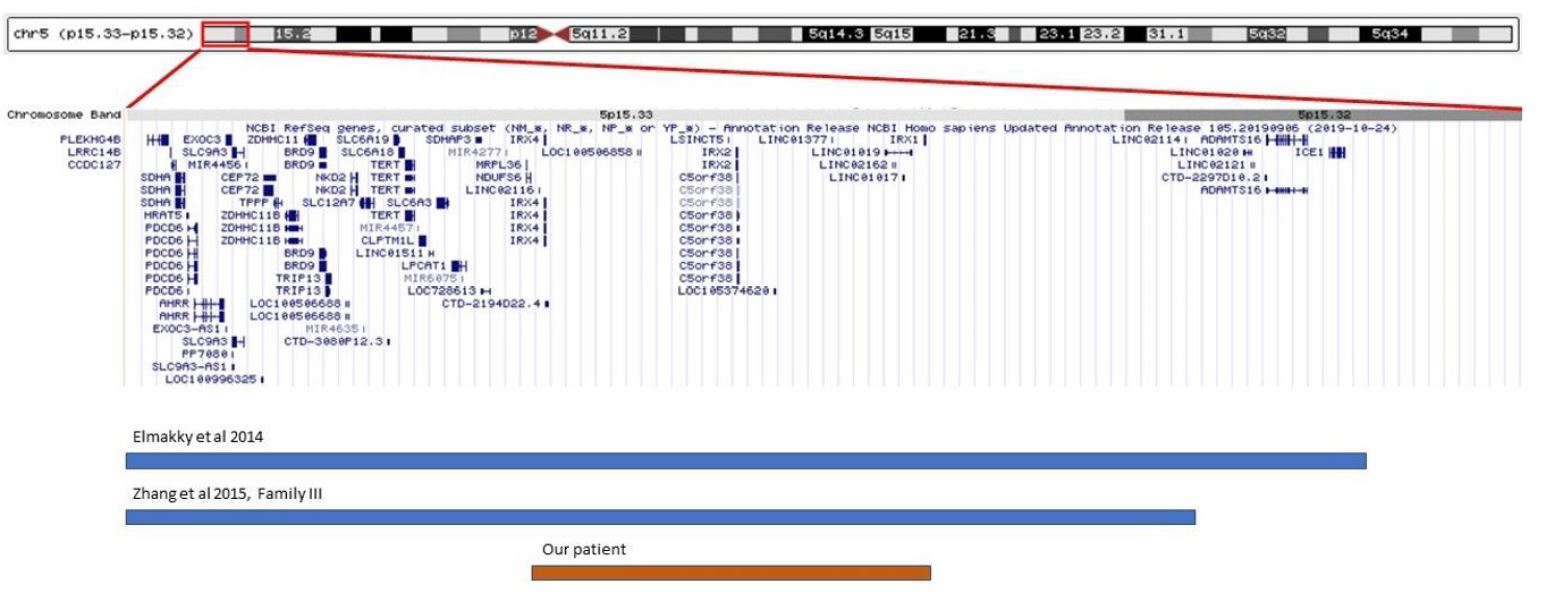
Most deletions on the short arm of chromosome 5 associated with Cri-du-chat syndrome are de novo deletions in the terminal region. Only 5% are interstitial, mostly with hereditary transmission.5 The deletion can vary in size from as little as ~5 Mb to the entire short arm of chromosome 5, and the larger the loss and the number of genes involved, the greater the likelihood of progression to global developmental delay.2,4,6 The corresponding phenotypes vary from minimal features to a complete phenotype. Several genotype-phenotype correlation studies have been published to identify the genomic regions responsible for the major features of this syndrome. However, the results have not always been consistent. In the case of the typical cat-like cry, recent studies have identified 5p15.31 (specifically the FLJ25076 gene), 5p15.32 (involving the ADAMTS16 and ICE1 genes), and 5p15.33 as possible critical regions.6,8,9,10
In this case, a smaller deletion than those described in the literature was detected, slightly less than 3 Mbp, located in the critical region of Cri-du-hat syndrome, but not involving genes previously considered essential for the presence of the cat-like cry. The region involved (5p15.33p15.32) is more distal than those originally associated with this trait.5,6 There are descriptions in the literature of two patients with small terminal deletions of 4.79 Mbp and 5.5 Mbp, both involving the region deleted in the present case (Figure 2).8,9 The patient with the 4.79 Mbp deletion did not show the typical cat-like cry, and the patient with the 5.5 Mbp deletion had this typical feature. The comparison of these two cases allowed to identify ADAMTS16 and ICE1 genes as possible critical genes in the development of the typical cat-like cry, but the present case seems to contradict this theory, since the observed deletion does not involve these two genes.8 The lack of reports of cases with identical size and location to this case makes it difficult to anticipate a prognosis for this patient. However, the location of the deletion in the critical region of Cri-du-chat syndrome suggests a possible etiology for the clinical signs described (high-pitched, cat-like cry associated with wide nasal bridge and thin upper lip). It should be noted that the first FISH study did not show any alterations because the deletion in question does not involve the FISH-targeted region, since the FISH probe is located in the telomere region. Considering the variability of the size of the deletion in Cri-du-chat syndrome and the variable location in 5p, aCGH is currently the primary diagnostic tool for this clinical entity, as it is able to precisely define the location and size of the deletions.3
It should also be noted that the parents of some children with this syndrome have the same deletion but do not show any characteristic features or symptoms, highlighting how environmental and/or genetic factors − such as incomplete penetrance or modifying genes − can influence the phenotypic expression.7 For this reason, the genetic study of parents is essential for genetic counseling. This was not done in the present case because the child had unknown parents, but this study would have been useful to assess the pathogenicity of the deletion in question, as there are no descriptions of identical deletions in the literature.
The study of associated malformations, namely cardiac, neurological, and renal, should not be neglected. Therefore, echocardiography and transfontanellar, abdominal, and renopelvic ultrasound should be performed.3)
Follow-up of this case did not reveal any additional features that could be part of the syndrome, and normal growth and development were noted.
The authors emphasize the rarity of this entity and the need for a high degree of clinical suspicion in its diagnosis, highlighting the importance of the diagnostic approach that allowed to clarify the etiology in the present case. This case also highlights the need for more information on atypical deletions such as the one considered here, in order to allow a more accurate and effective genotype-phenotype correlation. Although there is no specific treatment for this syndrome, its early recognition allows for timely and more effective educational and rehabilitation intervention. This case highlights the challenge that the diagnosis of a rare genetic disease can be and the importance of valuing clinical signs and performing genetic study, namely through chromosomal microarray analysis.