











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Spinal muscular atrophy (SMA) is one of the most common autosomal recessive diseases.1,2 It is the leading monogenic cause of death in infancy.3 The disease is caused by variants in the survival motor neuron (SMN) 1 gene on chromosome 5q13 that result in the absence of a functional SMN protein.1,2,4-8 This results in the degeneration and loss of alpha motor neurons in the brainstem and anterior horn of the spinal cord.1,2,4-8) The SMN2 gene is also capable of producing functional SMN protein, but at much lower levels because most mRNA transcripts do not include exon 7.1,3,7,9 Consequently, disease presentation may be modified by the number of SMN2 gene copies, with lower numbers associated with worse phenotypes.1,7,8) However, this genotype-phenotype correlation is not absolute.2
The disease phenotype is broad, leading to its classification into groups according to age at symptom onset and motor milestones achieved.3) Earlier-onset types are associated with a more rapidly progressive disease.1 This article will focus on type 1 SMA (SMA1), the most common form, which includes patients who present before 6 months of age with severe weakness and never achieve the ability to sit independently.1-3,10
The diagnosis of SMA1 should be suspected in infants with unexplained hypotonia and is established by molecular genetic testing.11 Nearly all patients have a homozygous deletion of SMN1 exon 7, with only about 5% having a single allelic deletion and a point mutation on the other allele.1,3,5 Regarding the number of SMN2 copies, in a large cohort of SMA1 patients, 86% were found to have two copies, 7% one copy, and 6% three SMN2 copies.2
Despite improvements in supportive care for SMA1 patients over the years, the prognosis remains poor.1 Disease progression leads to rapid weakness with respiratory failure and death before two years of age in most patients.1,2 Recently, therapies have been developed that lead to increased SMN protein production, improving motor function and survival, thus changing the natural history of the disease.1 In several countries, these therapies are initiated after clinical diagnosis has been established.1 The current literature supports the effectiveness and safety of these medications in pre/pauci-symptomatic stages of the disease.4,12,13 In order to identify and treat SMA1 patients early and to optimize therapeutic effects, some countries have implemented newborn screening (NBS) programs.5
Objectives
The aim of this study was to review the available literature on the importance of newborn screening and pre/pauci-symptomatic treatment of SMA1.
Main text
Clinical presentation
In SMA1 patients, there is a rapid and irreversible loss of motor neurons that begins in late fetal life and progresses during the first three months of life before the disease becomes clinically apparent, with 95% of motor neurons lost before six months of age.3,4,6,14 Clinically, there is also a rapid decline, with a mean age at symptom onset of 2.5 months (range 1.6-6 months).1,4,15,16 Recent studies suggest that 44% may present with symptoms in the first four weeks of life.17 At this stage, some patients may present with minor isolated signs, such as mild truncal hypotonia or weak reflexes, which may not be apparent on routine well-child examination.14,17,18) These subtle changes can be detected on examination by an experienced pediatric neurologist, with recent literature describing these patients as pauci-symptomatic.14
Patients present with weakness, hypotonia, and delayed or absent achievement of early motor milestones with an inability to ever achieve a sitting position.1 Neurologic examination reveals diffuse proximal predominant muscle weakness with decreased muscle tone and absent deep tendon reflexes.1,2) This weakness is more pronounced in the lower limbs.16 Reports on the natural history of SMA1 have shown that no patient achieved any level of sitting or head control by 12 months of age.16,19 Respiratory muscle weakness occurs early in the course of the disease with a weak cry and cough, paradoxical breathing, and the development of a characteristic bell-shaped chest.1,11) Bulbar involvement is common early and manifests as tongue fasciculations and poor swallowing reflexes with failure to thrive and increased risk of aspiration.1,11) Consequently, given the development of respiratory failure and swallowing dysfunction, respiratory and nutritional support is required in the first year of life.1) The cardiac muscle does not appear to be affected.11) Even with adequate supportive therapy, these patients were expected to die at a mean age of 13.5 months.1,11
Motor function scales have been adapted to monitor disease progression and measure disease activity.1 The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) scores motor activity on a 16-item scale from 0 to 4 (maximum score of 64) and is validated in newborns and infants with SMA1.1,20 The Hammersmith Infant Neurological Examination Section 2 (HINE-2) is an eight-item scale also used to evaluate infants with SMA1.1,20) Electrophysiologic studies can also be used to assess the extent of muscle denervation and measure the amount of intact motor neurons.1
Therapies
For a long time, supportive care was the only therapy available for SMA1 patients.3 Recently, three new disease-modifying therapies targeting SMN gene expression have been approved and shown to result in significant increases in motor scores.1,4,5,18) Until recently, these therapies were initiated after clinical diagnosis and genetic confirmation in many countries, including Portugal.3,4 The Portuguese NBS pilot study is ongoing since October 2022.
Onasemnogene abeparvovec (known as Zolgensma®) is a gene therapy using a recombinant adeno-associated virus (AAV9) vector encoding a functional SMN gene.8,20 It is administered intravenously as a single weight-based dose in children under five years of age.8) The Phase 1 START study enrolled 15 SMA1 patients, which were divided into a low-dose (three patients) and high-dose (12 patients) cohort, with a mean age at treatment of 6.3 months.21) In both cohorts, no patient required permanent mechanical ventilation at 20 months of age, exceeding previously reported ventilation-free survival time. At study cut-off, both groups had increased CHOP-INTEND scores from baseline, with more significant improvements in the high-dose group. At that time, 11 patients in this second cohort were able to swallow independently. Five-year follow-up showed a sustained therapeutic response with no regression of acquired motor milestones.22) At this time, two patients in the low-dose cohort were off mechanical ventilation and no patient in the high-dose cohort required permanent mechanical ventilation.
A Phase I/II study that included 12 symptomatic patients showed an improvement in CHOP-INTEND scores in all patients following early dosing with onasemnogene abeparvovec.13 However, a greater effect was observed in patients treated earlier (mean age of 1.8 months), with a mean increase of 13.7 compared to a 7.3 increase in those treated later (mean age of 5.1 months). At the 24-month follow-up, both groups had similar CHOP-INTEND scores despite significantly lower baseline scores in the early-treatment group. Only one patient in this study did not reach the unsupported sitting milestone, and this was the oldest child enrolled. In contrast, the two patients who were able to stand unassisted were both treated early and had baseline CHOP-INTEND scores of around 50. These results led to the conclusion that early treatment resulted in more significant improvements in motor function and higher CHOP-INTEND scores, regardless of disease severity.
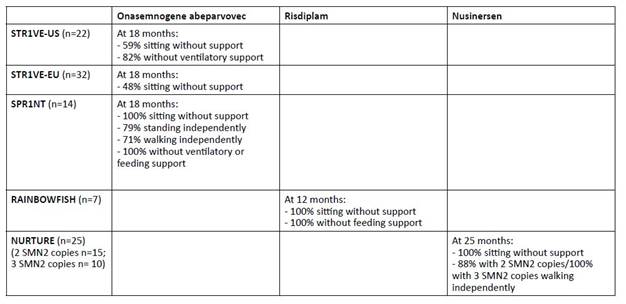
Two Phase III clinical trials in SMA1 patients younger than six months with two copies of SMN2, STR1VE-US and STR1VE-EU showed clinically significant efficacy.9,23 The United States (US) trial enrolled 22 patients. Of these, 13 (59%) were able to sit independently at 18 months, 20 (91%) were off mechanical ventilation at 14 months, and 18 (82%) were off mechanical ventilation at 18 months.9 In this study, 21 (95%) patients achieved a CHOP-INTEND score greater than 40. Historical cohorts have shown that untreated SMA1 patients have never been able to achieve or maintain a score greater than 40. The European (EU) study enrolled 32 patients and found that 31 (97%) did not require permanent ventilation by 14 months of age and 16 (48%) were able to sit unassisted by 18 months of age.23 One patient reached the milestone of standing with assistance and another was able to walk unassisted by 18 months of age.23) CHOP-INTEND scores greater than 40 were achieved in 73% of patients.
In the SPR1NT trial, 14 presymptomatic SMA1 infants with two copies of SMN2 were treated before six weeks of age.(24) Patients were referred either through prenatal screening or NBS (median age at diagnosis of 8 days and median baseline CHOP-INTEND of 49). All patients received onasemnogene abeparvovec at a median age of 21 days. These patients achieved unprecedented clinical improvement by 18 months of age: all patients were able to sit independently (79% within a normal developmental window), 79% were able to stand independently (50% within a normal developmental window), and 71% were able to walk independently (43% within a normal developmental window). All achieved CHOP-INTEND scores greater than 40. None of these children required any type of mechanical ventilation or feeding assistance at 18 months.
Side effects associated with the administration of onasemnogene abeparvovec have been reported. The most common have been pyrexia, thrombocytopenia without bleeding, and asymptomatic elevations in serum aminotransferase levels, mostly without hepatic dysfunction.1,9,20,21,23 Thrombotic microangiopathy is a rare but severe side effect.20) Others include asymptomatic elevations in troponin I levels without impact on cardiac function.1 Current treatment regimens include the use of oral prednisolone for 30 days with dose tapering in the following month if aminotransferase levels remain normal.1,6,20) Baseline laboratory testing is required, including AAV9 antibody titers.1,20 Positive titers (>1:50) complicate treatment.1,20
Risdiplam is a small molecule that modifies the splicing of SMN2 mRNA, increasing its efficiency and resulting in higher levels of SMN protein.1,20 It is administered orally once daily and is approved for patients with SMA two months of age and older.1,20 The multicenter FIREFISH study is a Phase II/III trial that enrolled 41 infants with SMA1 and two copies of SMN2.(25,26) The study was divided into two parts: Part 1, which has been completed, focused on selecting the dose for Part 2 and evaluating safety, pharmacokinetics, and pharmacodynamics; Part 2 evaluated the efficacy of the dose determined in Part 1.25,26 In this study, 29% of patients were able to sit unsupported for at least five seconds at 12 months.26,27 Regarding the CHOP-INTEND motor score at 12 months, 90% of patients achieved an increase of at least four points, with a median change from baseline of 20 points, and 56% achieved a score of 40 or higher.26,27 As noted above, historically, SMA1 patients have not been able to achieve such scores without treatment. At 12 months, 89% of patients were able to feed orally and 88% were alive without mechanical ventilation.27
RAINBOWFISH is an ongoing multicenter study evaluating the use of risdiplam in 18 pre-symptomatic SMA1 patients.28,29 The median age at first dose was 26.5 days and treatment was continued for a median of 8.7 months. Seven infants were treated for more than 12 months, all of whom were able to sit without support, maintained the ability to swallow solid food, and were able to feed exclusively by mouth at 12 months. Most of these patients achieved motor milestones within a normal developmental window.
No serious adverse events leading to risdiplam discontinuation have been reported in current studies.26,28 The most commonly reported adverse events were nasal congestion, cough, vomiting, diarrhea, pyrexia, and rash.1,28
Nusinersen, an antisense oligonucleotide designed to improve SMN protein expression via the SMN2 gene, was the first drug approved for the disease.3,8,20 It is administered intrathecally and requires a maintenance dose every four months.1,8,20 A multicenter study of symptomatic SMA1 patients under seven months of age showed a motor milestone response in 51% of those treated with nusinersen compared to no improvement in the control group.30 The treatment group also had a 47% lower risk of death or need for permanent ventilation.30) In this study, all improvements achieved with treatment were more pronounced when treatment was initiated earlier in the disease course.30) Further follow-up showed greater improvements in CHOP INTEND scores for those treated before 5.42 months, with 60% of these patients able to sit unassisted compared to only 38% of those treated later.8 In addition, 10% of the early-treated patients were able to walk with assistance, while none of the later-treated patients reached this milestone.8 Other studies have confirmed these findings, with more pronounced improvements in CHOP-INTEND motor scores for those treated at earlier ages.8
The NURTURE study, which investigated the use of nusinersen in presymptomatic patients six weeks of age or younger with two or three copies of the SMN2 gene, showed significant improvements, with 100% survival without the need for mechanical ventilation at 25 months of age.12 Additionally, all patients were able to sit independently and 88% of patients with two SMN2 copies were able to walk independently.12 All patients with three SMN2 copies were able to walk independently and achieved a maximum CHOP INTEND score of 64.12 Three (20%) patients with two SMN2 copies required nutritional support through a gastrostomy tube.12 These results are significantly different not only from the natural history of the disease, but also from those seen in patients treated in a post-symptomatic period.
To date, adverse events associated with nusinersen have been related to intrathecal administration and include headache or pain at the injection site.1,20
Key data from the above clinical trials are summarized in Table 1.
Based on the currently available data, algorithms in several countries where NBS programs are available recommend immediate treatment for those diagnosed through NBS programs with two, three, or four copies of SMN2 to maximize treatment response.3,31
Newborn screening
Evidence has clearly shown that treatment at a pre-symptomatic stage is necessary for maximum therapeutic benefit. This has led to the development of newborn screening (NBS) programs aimed at identifying pre-symptomatic newborns. These are highly feasible, as the majority of patients have a single common pathogenic variant, and have been implemented in several countries.4,5,7,32) Real-time polymerase chain reaction (PCR), which detects SMN1 from a dried blood spot, appears to be inexpensive and has a positive predictive value of 100%.3,4) Currently, only about 2% of the world’s population is screened.5
Data from countries where NBS has been implemented show that it is extremely reliable.5 The false positive rate is very low (lower than for other diseases already included in NBS programs) and is related to either low white blood cell counts or earlier versions of PCR primers.5) False negatives have not been identified.5) Recently published experience from New York State found that results were reported at a median of seven days of life and that the first evaluation occurred at a median of nine days of life.33 No false-positive results were found.33 Most infants started treatment before six weeks of age.33 Delays in treatment were most commonly due to the presence of AAV9 antibodies or elevated troponin I levels, or were insurance-related.33
The Portuguese NBS program was created in 1979 with the aim of identifying rare, treatable diseases at a pre-symptomatic stage and allowing the implementation of appropriate treatment.34 Given its well-developed operational structure and a national coverage rate close to 100%, the inclusion of SMA in this program seems to be the best option for early diagnosis. Since October 2022, a pilot study is underway to evaluate the technical and organizational feasibility of SMA screening in Portugal and to determine its public health impact. This study aims to screen 100,000 newborns over a period of up to two years.35
Conclusions
Historically, SMA1 patients had rapid symptom progression, never attained the ability to sit independently, and typically died of respiratory failure before the age of two. More recently, disease-modifying therapies have been developed and marketed. These have proven to be relatively safe and effective. However, to achieve maximum benefit, these treatments should be initiated in pre-symptomatic patients.
In most countries where NBS programs have not been implemented, diagnosis of SMA1 is typically delayed and the majority of motor neurons have already been lost. At this stage, disease-modifying therapies do not provide maximum benefit. However, treatment of pre-symptomatic patients is not only safe, but also effective in altering the natural course of the disease. Because these patients require less ventilatory and nutritional support, early treatment also has the added benefit of reducing the overall cost of therapy.
The only way to ensure early treatment is to identify patients at a pre-symptomatic stage through NBS programs. Real-world data from countries where these programs have been implemented have shown them to be highly reliable.
By reviewing the current evidence, this article has demonstrated the benefits of implementing NBS programs on the quality of life of patients and their families. A pilot study to evaluate the implementation of an NBS program for SMA1 is currently underway in Portugal.

