











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Neuroblastoma is the most common extracranial solid tumour in the paediatric population, accounting for approximately 8-10% of all childhood cancers.1,2
The tumour arises from the abnormal proliferation of embryonic neural crest cells which normally give rise to the sympathetic nervous system (SNS) and the adrenal gland medulla. Clinical presentation depends on the primary tumour location and the presence of metastatic disease.3
Neuroblastomas have a uniquely heterogeneous course, which distinguishes them from other solid tumours. The clinical evolution ranges from spontaneous regression to widespread metastatic disease with limited treatment response, despite aggressive multimodal therapy including induction chemotherapy, surgery, radiotherapy, high-dose chemotherapy with autologous stem cell rescue, and biologic and immunotherapeutic maintenance therapy.2
The treatment of choice depends on risk stratification based on disease stage, patient age at diagnosis and tumour biological characteristics.2
Clinical case
We present the case of an eight-month-old male infant, born prematurely at 35 weeks from a dichorionic diamniotic twin pregnancy, with no other relevant personal or family history.
At the age of eight months, his mother reported a perceived increase in abdominal volume over the previous month, with no accompanying symptoms such as fever, weight loss, perceived abdominal discomfort, changes in gastrointestinal transit, vomiting, respiratory distress or skin lesions. Physical examination revealed a palpable hepatic edge about 10cm below the right costal margin.
Abdominal sonography (Fig. 1) confirmed hepatomegaly (10,6cm) and numerous nodular formations suggestive of metastatic lesions, the largest measuring 21mm. No other ultrasound abnormalities were detected, namely space-occupying lesions in the adrenal gland areas or along the paravertebral space. Chest radiography was normal.
Laboratory findings showed only increased aminotransferases (AST 79 UI/L; ALT 74 UI/L) and lactate dehydrogenase (LDH 345 UI/L). The remaining laboratory data, including hemogram, renal function, serum electrolytes, uric acid, and inflammatory parameters, were within normal limits. Viral serologies were negative for Epstein-Barr, Varicella-zoster, Hepatitis B and C and Human Immunodeficiency virus.
Vanilmandelic acid/creatinine (VMA/Cr) ratio and neuron-specific enolase (NSE) were above the upper limits of normal (ULN): VMA/Cr 80,1mg/g (ULN 2-12mg/g); NSE 55,6ng/mL (ULN <18.3ng/mL). The I-123 metaiodobenzylguanidine (mIBG) scan (Fig. 2) revealed intense liver uptake but no evident primary lesion location. However, increased uptake was observed on the left dorsolombar paravertebral region, which could correspond to physiological adrenal gland uptake.
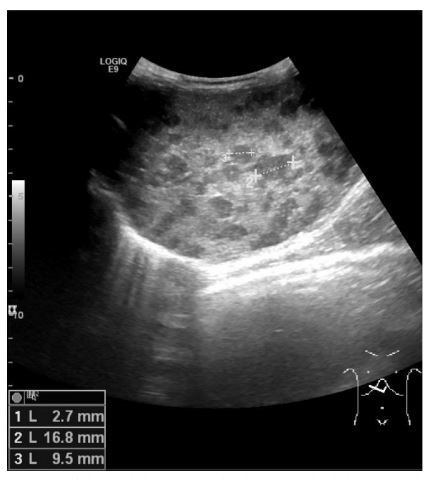
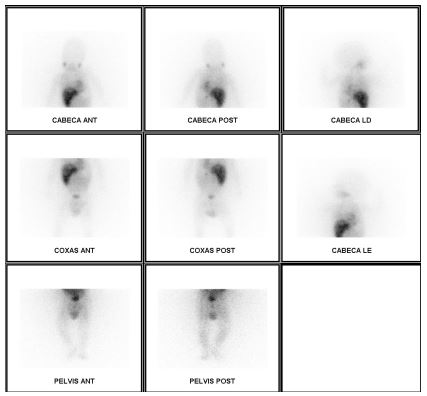
Figure 2 mIBG scan with intense 123 I-mIBG liver uptake, especially in the left dorsolombar paravertebral region, evidencing adrenal gland physiologic uptake
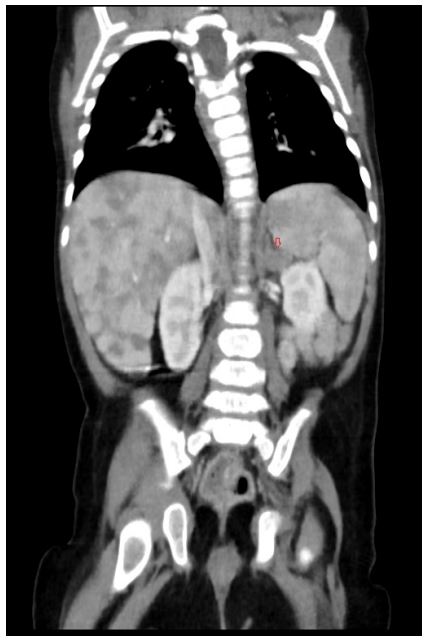
Figure 3 CT-scan showing multiple hepatic metastases and increased left adrenal volume (arrow) suggesting a primary tumour arising from the left adrenal gland
Thoracoabdominal computed tomography (CT) (Fig. 3) showed an enlarged left adrenal gland (15mm), along with the previously identified hepatic lesions, suggesting a primary tumour arising from the left adrenal gland with secondary liver metastasis. Bone marrow aspirate and biopsy (performed for staging) showed no abnormalities.
Based on these findings, a diagnosis of neuroblastoma, stage MS in the INRGSS classification was assumed.
Since the primary lesion was only inferred from the adrenal gland enlargement and a liver biopsy could pose significant iatrogenic risks to the patient, a multidisciplinary team opted for a “wait-and-see” approach with close monitoring.
The standard follow-up in these patients includes clinical examination with appropriate radiological investigations every eight weeks until the tumour regression begins, then every 12 weeks for one year and then annually for five years.
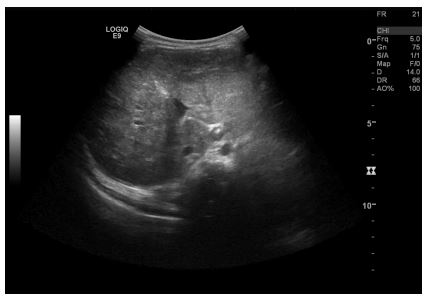
In this case, a more frequent follow-up schedule was chosen: an appointment every four weeks until a reduction in liver lesions size was observed, involving a clinical, laboratory and ultrasound evaluation. Clinically, there was a progressive decrease in abdominal circumference, with no other symptoms reported. Laboratory follow-up revealed a favourable course, with normalization of aminotransferase, catecholamine excretion and liver size after eleven months (table 1). Radiologic follow-up was performed by abdominal ultrasound, revealing a normalisation of adrenal gland and liver dimensions three and nine months post-diagnosis, along with progressive involution of the hepatic lesions (Fig. 4). By the 11-month follow-up, a normal abdominal ultrasound was reported.
Table 1 AVM/Cr and NSE evolution
| Months after diagnosis | VMA/Cr (mg/g) | NSE (ng/mL) |
| 0 | 80.1 | 55.6 |
| 1 | 31.2 | 44.4 |
| 2 | 29.9 | 26.9 |
| 3 | 19.5 | 17.9 |
| 11 | 12 | 15.9 |
Discussion/conclusions
Neuroblastoma is the most common extracranial solid cancer in infancy, with a median age at diagnosis of 18 months.1,4
Clinical manifestations vary according to the primary tumour location and the presence of metastatic disease2. The most common primary site of neuroblastoma is the adrenal gland, as in the present case, although it can occur anywhere along the SNS. Abdominal masses, the most common presentation, may be asymptomatic and detected incidentally during a routine medical assessment, as an increased abdominal volume noticed by the parents or as a radiological finding. Alternatively, it may result in abdominal pain, distention, constipation, hypertension and dyspnea due to local effects on abdominal and thoracic organs. Massive abdominal masses are oncologic emergencies, as they can lead to life-threatening complications such as respiratory impairment or abdominal compartment syndrome.1-4
Metastatic disease is found in approximately 50% of newly diagnosed patients, with the most common metastatic sites being the the bone, bone marrow, liver and skin.1,4
Diagnosis is suggested by radiologic and laboratory findings and is confirmed by histological analysis of the primary or metastatic lesions on biopsy.1,2
In the present case, a normal hemogram suggested the absence of bone marrow involvement. Increased aminotransferases were associated with the presence of liver metastasis.1
Urinary catecholamine metabolites, VMA and homovalinic acid (HVA), are secreted by neuroblastoma cells and are elevated in 90% to 95% of patients.1,3 They are also useful for disease monitoring.3
NSE is not specific for neuroblastomas, however, increased levels (> 100 ng/mL) correlate with advanced-stage disease and a poor survival rate.3
Although abdominal ultrasonography and chest radiography are commonly performed, the International Neuroblastoma Risk Group (INRG) recommends CT or magnetic resonance imaging (MRI) to assess primary tumour size, regional invasion, lymph node involvement and distant metastasis.1,4
In our patient, a CT scan was performed considering the young age and the risks associated with sedation, as it can usually be conducted without sedation while still achieving a technical satisfactory examination. The CT scan was very important not only for confirming liver metastasis but also for suggesting the primary tumour location.
The radiolabeled mIGB scan uses a norepinephrine analogue (mIGB) which is highly specific and sensitive for detecting bone disease, as mIGB is taken up by neuroblastoma cells in 90% of the cases.1,4
Bone marrow aspirate and biopsy are important staging exams. In cases where bone marrow involvement is detected and biopsy of other sites is not feasible, they can also be useful for diagnosis confirmation and the study of histologic and biologic tumour characteristics, which are essential for risk stratification.2
Given the wide variability in neuroblastoma behaviour, treatment choice is based on the risk of aggressive disease. The International Neuroblastoma Risk Group Staging System (INRGSS) classifies pre-surgical tumour staging into four levels, based on disease extent and the presence of image-defined risk factors (IDRF).5
Risk stratification is determined by tumour staging, patient age at diagnosis, tumour histology and grade differentiation, and molecular alterations.6,7
The eight-month-old patient presented with metastatic disease limited to the liver. Based on the characteristics described, we classified the tumour as stage MS. Patients with stage MS neuroblastoma belong to the low-risk group: ≤ 12 months of age, MYCN non-amplified neuroblastoma, without segmental chromosomal alterations or life-threatening symptoms. These infants can have disseminated disease involving the skin, liver and/or bone marrow but no involvement of the bone, pleura, lung or CNS. MS neuroblastoma is an exception to the generally poor prognosis of children presenting with metastatic disease, as the tumour can spontaneously regress without treatment, as happened in this case.4-10
Bone marrow aspirate and biopsy did not detect tumour cells. To evaluate the tumour’s histologic and biologic characteristics, a biopsy of either the primary tumour or metastatic lesions would be required. Among biological markers, oncogene MYCN amplification is the most significant, occurring in approximately 20% of neuroblastomas and correlating to advanced-stage disease, rapid progression and poor clinical outcome. The presence of segmental chromosomal alterations is also a worse prognosis warning. Conversely, a genomic profile characterised by whole chromosome gains or losses is more frequently observed in localised tumours and in children under 12 months, indicating a better prognosis.11
Histology and genetic characterization are important for staging. Given the risks associated with an open surgical biopsy (as percutaneous biopsy is not recommended for neuroblastoma diagnosis), the patient’s young age, and the favorable clinical presentation, our team decided to establish the neuroblastoma diagnosis based on biomarkers, mIBG and age. The multidisciplinary team (Pediatric, Radiology and Pediatric Surgery Departments) decided to do a “wait-and-see approach”. This decision was supported by data from the German Society for Paediatric Oncology and Haematology (GPOH group).12
De Bernardi reported an overall survival rate of 82% for infants with MS stage neuroblastoma and under one year of age, compared with 31% for children over one year o. Infants under six months had a better prognosis than those aged six to 12 months (86% vs 78%). Disease progression to stage M occurred in only 1-2% of the cases. The National Cancer Institute supports the idea that children up to six months of age may not require a biopsy or surgical removal of the tumour as it may regress without treatment.13
The decision to adopt a wait-and-see approach required close surveillance, with sequential clinical observations, laboratory tests and abdominal ultrasonography. This strategy allowed for a less invasive, less iatrogenic approach while ensuring patient safety.

