










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.31 no.5 Coimbra out. 2013
https://doi.org/10.4152/pea.201305257
Application of Ti/Pt/β bbbβ-PbO2 Anodes in the Degradation of DR80 Azo Dye
J. Florêncio, M.J. Pacheco, A. Lopes and L. Ciríaco*
UMTP and Department of Chemistry, University of Beira Interior, 6201-001 Covilhã, Portugal
Abstract
This study describes the application of a Ti/Pt/PbO2 electrode as photoanode in the degradation of the dye Direct Red 80 (DR80). The electrode was prepared by platinization of a titanium substrate, followed by electrodeposition of a PbO2 layer. The presence of β-PbO2 phase was confirmed by X-ray diffraction. DR80 degradation tests were performed by different techniques, namely, photolysis, photocatalysis, electrocatalysis and photoelectrocatalysis. The best colour removals were obtained in the photoelectrocatalysis assays and were higher than 85% for 5.0 and 12.5 mg L-1 dye initial concentration and about 72% for 25.0 mg L-1 DR80 initial concentration, after applying a current intensity of 5 mA for 6 hours. At a DR80 initial concentration of 25.0 mg L-1 , the best absorbance removals were obtained in the photoelectrocatalytic assays, with a current intensity of 50 mA, which led to a colour removal of 100% after 4 hours. Photolysis and photocatalysis presented similar colour removals that were 62, 26 and 18% for the initial concentrations of 5, 12.5 and 25 mg L-1, respectively.
Keywords: Ti/Pt/β-PbO2 anode; anodic oxidation; DR80; photoelectrocatalysis.
Introduction
The untreated effluents from several industries, such as textile, printing paper, food processing and cosmetic manufacturing, present high level of pollutants and colour. Essentially, this is due to the presence of dyes, most of them from the azo dyes group, with the general formula R1-N = N-R2 [1-3]. Therefore, effluents from those industries are responsible for the dissemination of these environmental pollutants. These dyes, besides contaminating the natural aquifer resources with persistent organic pollutants, which may be toxic to aquatic fauna and flora, also give them colour, thus preventing light penetration and natural photosynthetic processes of the native organisms.
A representative azo dye, possessing 4 azo bonds, is the direct red dye DR80. Its degradation was already performed using non-biological processes, such as anodic and photocatalytic oxidation, with good results [3,4]. For the complex effluents containing dyes, these advanced oxidation processes (AOPs) can be a suitable alternative to conventional technologies. In fact, these processes are capable of producing highly oxidizing species, like hydroxyl radicals, and can be an efficient treatment for waters and wastewaters. There are various techniques among the AOPs for the processing of contaminated waters [4-12], and anodic oxidation can be an efficient option for pollutant's degradation [3,5,6,11-22]. However, the performance of the anode depends strongly on the electrode material [3,5,13-19] as well as on the experimental conditions [20-22]. Many electrode materials have been tested: noble metals, different forms of carbon, boron-doped diamond (BDD) and metal oxides. In the class of metal oxides, PbO2 is among those already tested as anodes for pollutant's oxidation [13-18]. Besides its ability as anode, PbO2 presents band gap energy of 1.4 eV that allows its application as photocatalyst. This anode has already been used with solar light with success in the photomineralization of oxalic acid and phenols [23,24] and with UV light (365 nm) in mineralization selectivity studies of different compounds [25]. In this latter study, its performance was compared with that of TiO2 and the authors have concluded that PbO2 can be used to photomineralise only some phenols, in contrast to TiO2 that does not show selectivity towards the different organic compounds.
In this work, the use of PbO2 as photocatalytic material for the degradation of solutions containing a model tetra azo dye, direct red 80, was tested, using UV light with a wavelength of 254 nm. In order to enhance the PbO2 catalytic effect, degradation assays were electrochemically assisted with low intensity currents.
Experimental
Chemicals
Ti/Pt/PbO2 electrodes were prepared from titanium foil (99.7%, Sigma-Aldrich), H2PtCl6 (99.9%, Sigma-Aldrich), HCl (Merck, 37%), sodium dodecyl sulphate (Sigma-Aldrich, 99%), HNO3 (65%, Merck) and Pb(NO3)2 (99%, Sigma-Aldrich).
The chemicals used as model pollutant and as electrolyte were DR80 (25%, Aldrich) and sodium sulfate (99%, Merck), respectively. The molecular structure of the DR80 is shown in Fig. 1.
Electrode preparation and characterization
The electrode preparation was carried out according to literature in three steps [15,16]: pretreatment of the titanium substrate, followed by platinization with H2PtCl6 and posterior electrodeposition of PbO2. The Ti/Pt/PbO2 electrode structure was characterized by X-ray powder diffraction (XRD), at room temperature, by means of a Rigaku DMAXIII/C diffractometer, using Cu K radiation (λ = 0.15406 nm) and working at 30 kV/40 mA. The diffraction patterns were collected between 2θ 20 and 80°, with a 0.02° step, and acquisition time of 2 s per step. The morphology of the prepared electrodes was characterized by scanning electron microscopy (SEM) with a Hitachi (S-2700)/Oxford (60-74) system operating at 20 keV.
The electrochemical characterization was performed by cyclic voltammetric measurements with a potentiostat/galvanostat VoltaLab PGZ 301, in a one- compartment cell, with a 4 cm2 Ti/Pt/PbO2 electrode as the working electrode, a 4 cm2 platinum plate as the counter electrode and a commercial saturated Ag/AgCl, KClsat electrode as the reference electrode. Voltammograms were recorded at scan rates between 10 and 100 mV s-1 in 0.035 M Na2SO4 aqueous solution without and with DR80 concentrations of 1 g L-1.
Degradation assays
The degradation of DR80 solutions was performed using aqueous solutions with different initial dye concentrations, namely 5.0, 12.5 and 25.0 mg L-1, containing Na2SO4 (5 g L-1) as background electrolyte. Four different techniques were used in the study, photolysis, photocatalysis, electrocatalysis and photoelectrocatalysis. In the photo-assisted assays, radiation was provided by an immersed UV lamp, of 7 W, that emits a radiation with a wavelength of 254 nm. All the assays were performed at 25 °C, in a thermostatized system with stirring, and the processed solution volume was 200 mL.
For all DR80 concentrations tested, the electrocatalysis and photoelectrocatalysis assays were performed applying a current intensity of 5 mA, using a potentiostat/galvanostat VoltaLab PGZ 301 and a one-compartment three-electrode cell. The anode was a Ti/Pt/PbO2 electrode and the cathode a stainless steel foil, being the geometric area of both electrodes 20 cm2.
For the initial concentration of 25 mg L-1, tests applying a current intensity of 50 mA were also performed.
The DR80 degradation was monitored by two techniques, UV-Vis absorption spectrophotometry, using a Shimatzu UV-1800 spectrophotometer, and total organic carbon (TOC) determinations, using a Shimadzu TOC-VCPH/CPN analyzer.
Results and discussion
Electrode characterization
Fig. 2a shows the X-ray diffractogram of the electrode surface.
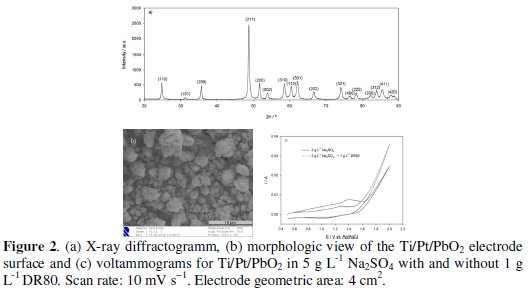
The diffraction lines were indexed according to JCPDS ICDS data (PDF #41-1492) and the presence of the β-PbO2 phase was confirmed. The morphology of the electrode surface is also presented in Fig. 2b, where a compact coating can be observed, with 1 μm granules, being some of them aggregated.
In Fig. 2c, voltammograms for the systems electrolyte and electrolyte+DR80 are presented. The two consecutive anodic peaks observed in both systems correspond to the partial oxidation of Pb2+ surface atoms to Pband then to Pb4+[26]. There are no other peaks besides these, showing that any DR80 oxidation reaction happens for potentials in the oxygen evolution zone (higher than 1.5 V vs. Ag|AgCl, KClsat). In the electrodegradation assays, the current intensities of 5 mA and 50 mA correspond to an applied potential of 1.5 V and 2.0 V vs. Ag|AgCl, KClsat, meaning that DR80 oxidation was performed in the oxygen evolution region, where HO• radicals can be formed. Fig. 2c also shows a decrease in the current intensity when the azo dye is added to the electrolyte solution that must be due to a partial passivation of the electrode surface for potential lower than the oxygen evolution potential.
Degradation assays
Fig. 3 to 5 present the spectrophotometric results obtained for the assays performed with initial DR80 concentrations of, respectively, 5.0, 12.5 and 25.0 mg L-1.
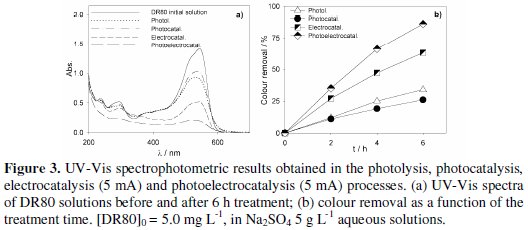
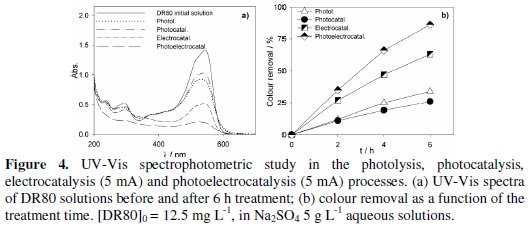
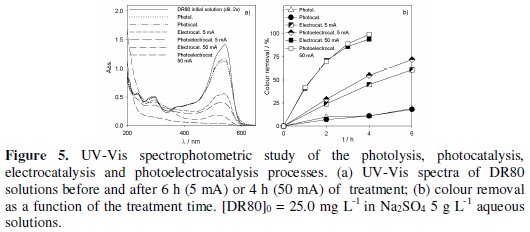
The absorption spectra of the DR80 initial solutions at the different concentrations used are shown in Fig. 3a, 4a and 5a. They are characterized by one band in the visible region, at about 538 nm, due to the absorption of the azo bond conjugated with the aromatic rings, and bands in the UV region, attributed to the absorptions of benzenic and naphthalenic rings [27]. By analyzing the spectra of the samples taken after 6 hours of treatment, for all the degradation processes studied, it can be seen that the decay in the absorbance at 538 nm is always faster than those in the UV region. This means that the azo bond, responsible for the absorption in the visible region, is the first to be broken. In the case of photoelectrocatalysis, after 6 hours of treatment, the spectra profile is different, since the DR80 characteristic bands almost disappear, which means that not only the azo bonds were broken but the opening of the aromatic rings also occurred.
For the lowest DR80 concentration studied (Fig. 3), after 6 hours degradation assays, 73% colour removal was obtained with photolysis and with electrocatalysis, and 88% with photoelectrocatalysis at 5 mA.
For DR80 initial concentration of 12.5 mg L-1 (Fig. 4), the relative order of colour removals did not change, i.e., the best results were also obtained in the photoelectrocatalysis (86%) and the lowest in the photocatalysis (26%). Actually, photocatalysis presented a poorer performance than photolysis. Apparently, the PbO2 film does not present photocatalytic activity for the radiation used and, in fact, it seems to prevent the dispersion of the radiation in the solution. In spite of the absorbance removal percentage results presented in Fig. 5 seem worst than those in Fig. 4, this is only due to the different initial DR80 concentrations used, since equal absorbance removals in percentage represents double absolute absorbance removal in the case of the assay performed with the highest concentration.
For the highest DR80 concentration studied, 25.0 mg L-1 (Fig. 5b), after 6 hours assay's duration the reductions in absorbance at 538 nm for the different applied processes were the following: photolysis/photocatalysis -18%; electrocatalysis at 5 mA -61%; photoelectrocatalysis at 5 mA -72%. However, when a 50 mA current intensity was imposed in the photoelectrocatalytic assay, instead of 5 mA, a faster decolourisation of the solution was observed and, after 4 h of treatment the colour removal was complete. For this applied current intensity, the absence of bands in the UV region of the spectrum presented in Fig. 5a proves that the benzenic and naftalenic rings opening occurs almost completely. With the applied current intensity of 5 mA, even after 6 hours there was still a residual colour. This difference in absorbance removals with the applied current intensity must be due to an indirect oxidation process, mediated by hydroxyl radicals, formed when the applied current exceeds the limiting current for the DR80 concentration under study, i.e., when the process is definitively controlled by mass transfer.
Fig. 6 presents a comparative study of DR80 degradation by photolysis, photocatalysis, electrocatalysis (5 mA) and photoelectrocatalysis (5 mA) after 6 hours of treatment, for the three DR80 initial concentrations studied.
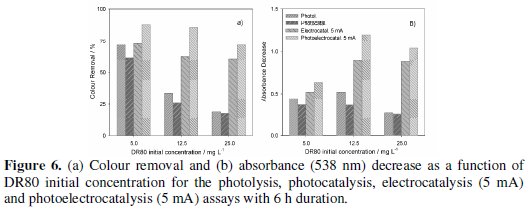
Experimental data show that for the lowest concentration, the UV irradiation can have a positive effect on the degradation of the dye and decolourisations of 72% were attained. However, when DR80 concentration increases to 25 mg L-1, the colour removal expressed in terms of percentage is very low, around 19%. But if the absolute absorbance removals are compared, as in Fig. 6b, it can be seen that this value does not vary too much with the initial concentration. Thus, for the highest DR80 initial concentration tested the UV irradiation contribution is low, being the application of an electrical current more favourable. In fact, in the photoassisted electrochemical process, even for the highest DR80 initial concentration, a 72% of colour removal was achieved.
In all the assays of photolysis and photocatalysis, for equal initial concentration similar results were attained, being the photocatalytic effect of the electrode not detected in this study, probably because the radiation used was not the most adequate to stimulate the PbO2 catalytic effect.
Regarding TOC removals, even after 6 hours of treatment the DR80 degradations by photolysis and photocatalysis do not present any significant mineralization. In Fig. 7 the TOC removals for electro and photoelectrocatalytic processes are presented.
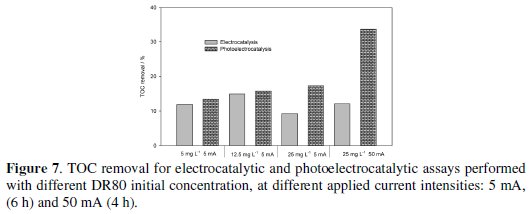
The best removals were always observed for the photoelectrocatalytic processes, and were 20% and 30%, for 6 h assay run at 5 mA and 4 h assay run at 50 mA, respectively. The absolute TOC removals increase with DR80 initial concentration, showing the importance of the mass transfer process.
Conclusions
From the comparative study of DR80 degradation by photolysis and photocatalysis, with PbO2 catalyst, and electrocatalysis and photoelectrocatalysis, using a PbO2 photoanode, the following conclusions can be drawn:
• The degradation of the DR80 dye molecules can be achieved using PbO2 photoanodes. The first step of the degradation mechanism must be the breaking of the azo bond, since the absorbance decreases during the processes without significant reduction in the TOC content of the solutions, meaning that mineralization is very poor.
• The photolysis effect is important for the lowest DR80 initial concentration studied, but for higher concentrations the effect of current intensity is predominant.
• The best colour removals, higher than 85%, were obtained in photoelectrocatalysis assays, with 5.0 and 12.5 mg L-1 DR80 initial concentration, at 5 mA, after 6 hours.
• For the DR80 initial concentration of 25.0 mg L-1, a complete colour removal was obtained in the photoelectrocatalysis tests, after 4 hours at a current intensity of 50 mA. The best TOC removal (30%) was also obtained for this assay.
References
1. Zollinger H. Color chemistry, synthesis. Properties and applications of organic dyes and pigments. New York: VCH Publishers; 1991. [ Links ]
2. Hao O J, Kim H, Chiang P-C. Crit Rev Environ Sci Technol. 2000;30:449-505. [ Links ]
3. Pacheco M J, Ciriaco M L F, Lopes A, et al. Port Electrochim Acta. 2006;24:273-282. [ Links ]
4. Mahmoodi N M, Arami M, Limaee N Y, et al. Chem Eng J. 2005;112:191-196. [ Links ]
5. Panizza M, Cerisola G. Electrochim Acta. 2003;48:3491-3497. [ Links ]
6. Panizza M, Cerisola G. Electrochim Acta. 2004;49:3221-3226. [ Links ]
7. Konstantinou I K, Albanis T A. Appl Catal B-Environ. 2004;49:1-14. [ Links ]
8. Akpan U G, Hameed B H. J Hazard Mater. 2009;170:520-529. [ Links ]
9. Soutsas K, Karayannis V, Poulios I, et al. Desalination. 2010;250:345-350. [ Links ]
10. Tehrani-Bagha A R, Mahmoodi N M, Menger F M. Desalination. 2010;260:34-38. [ Links ]
11. Kariyajjanavar P, Jogttappa N, Nayaka Y A. J Hazard Mater. 2011;190:952-961. [ Links ]
12. Raghu S, Lee C W, Chellammal S, et al. J Hazard Mater. 2009;171:748-754. [ Links ]
13. Ciriaco L, Anjo C, Correia J, et al. Electrochim Acta. 2009;54:1464-1472. [ Links ]
14. Martinez-Huitle C A, Quiroz M A, Comninellis C, et al. Electrochim Acta 2004;50:949-956. [ Links ]
15. Andrade L S, Ruotolo L A M, Rocha-Filho R M, et al. Chemosphere. 2007;66:2035-2043. [ Links ]
16. Andrade L S, Tasso T T, Silva D L, et al. Electrochim Acta. 2009;54:2024-2030. [ Links ]
17. Hmani E, Samet Y, Abdelhedi R. Diam Relat Mat. 2012;30:1-8. [ Links ]
18. Aquino J M, Pereira G F, Rocha-Filho R C, et al. J Hazard Mat. 2011;192:1275-1282. [ Links ]
19. Zhou M, Sarkka H, Sillanpaa M, Sep. Purif. Technol. 2011;78:290-297. [ Links ]
20. Aquino J M, Rodrigo M A, Rocha-Filha R C, et al. Chem Eng J. 2012;184:221-227. [ Links ]
21. Cruz-Gonzalez K, Torres-Lopez O, Garcia-Leon A M, et al. Desalination 2012;286:63-68. [ Links ]
22. Tsantaki E, Velegraki T, Katsaounis A, et al. J Hazard Mat. 2012;207:91-96. [ Links ]
23. Karunakaran C, Dhanalakshmi R. Sol Energy Mater Sol Cells. 2008;92:588-593. [ Links ]
24. Karunakaran C, Dhanalakshmi R. Sol Energy Mater Sol Cells. 2008;92:1315-1321. [ Links ]
25. Karunakaran C, Dhanalakshmi R. Cent Eur J Chem. 2009;7:134-137. [ Links ]
26. Songa S, Zhana L, Hea Z, et al. J Hazard Mat. 2010;175:614-621. [ Links ]
27. Silverstein R, Bassler G, Morril T. Spectrometric identification of organic compounds. 4th ed. New York: Jonh Wiley and Sons, Inc.; 1981. [ Links ]
*Corresponding author. E-mail address: lciriaco@ubi.pt
Received 12 July 2013; accepted 31 October 2013

