











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
The iron concentration determination in water resources is of great interest. Iron is an essential element for biological processes in living organisms. It is one of the common elements in drinking water resources. Usually, the iron (II) concentration in anaerobic groundwater resources is 0.5-10 ppm [1]. In most countries, the considered iron concentration in drinking water is less than 0.3 ppm [1]. Iron exists in water mainly as iron (II) or iron (III) state. Iron (II) is a water-soluble species and rapidly converts to iron (III) in an oxygen-rich environment [3]. Various analytical methods have been developed for iron determination in an aqueous medium, such as spectrophotometry [3, 4], inductive coupled plasma optical emission spectrometry (ICP-OES) [5, 6], atomic absorption spectrometry (AAS) [7, 8], voltammetry [9-14] and chromatography [15, 16]. Most of these methods are expensive and require complex analytical instruments, while the recent trend in chemical analysis is developing green alternative analytical methods with simple methodologies and instrumentations. Electrochemical analysis methods tune extensively with these principles.
Solid electrodes have been widely applied in electrochemical analysis processes, because of their ease of reproduction, their negligible toxicity [17], and the significant ability to modify their surface [18].
Iodine-coated polycrystalline platinum electrode demonstrated to be a model for the highly reactive platinum electrode surface modification. The adsorbed iodine converts the highly reactive platinum electrode surface into a passive surface [19]. Subsequently, the adsorbed iodine monolayer stays stable, even if the electrode is taken out of the solution or rinsed extensively with an iodine-free aqueous solution [20]. Carbon monoxide generation causes the iodine adatoms elimination from the iodine-coated platinum electrode surface [21]. Also, the scanning beyond the potential window (-0.2 - 0.8 V) in both directions desorbs iodine from the electrode surface [22, 23].
The developed iodine-coated platinum electrode, with and without modification, was used for the quantitative determination of some organic and inorganic species in aqueous solutions [19, 24-27] and in biological tissue [28].
The easiness of modified iodine-coated platinum electrode preparation is combined with electrochemical methods benefits such as simplicity, reduction in chemical reagents needs and rapidity. The aim of this study was utilizing the sensitivity and selectivity of anodic stripping voltammetry to develop an analytical method for iron determination in groundwater resources.
Experimental
Instruments, cell and materials
A PAR potentiostat (model 362, EG & G) interfaced with a computer via GPIB interface (IEEE), for potential control and data acquisition, was used. Locally modified Labview® (IEEE) software was used for data acquisition. A home-made H-shape electrochemical cell with multiple inlet/outlet for gas purging and blanketing was used. All solutions were degassed before every experiment, and the measurements were carried out under a nitrogen atmosphere. Also, the solution delivery to and out of the cell was used for voltammetric applications. This electrochemical cell was composed mainly of two asymmetrical compartments separated by a sintered glass frit to avoid the diffusion of undesirable species towards the working electrode compartment. The working electrode was housed in one compartment, while the reference and the auxiliary electrodes were housed in the other compartment. The working electrode was a 0.5 mm polycrystalline platinum wire purchased from Aldrich (99.99% minimum purity certified reagent). The immersed end of the platinum electrode was curved like a U-shape to give a mark under the solution surface, for achieving a consistent surface area. The reference electrode was a home-made silver/silver chloride in a 1.0 M KCl solution. The auxiliary electrode was a spiral-shaped platinum wire (Aldrich, certified 99.99% minimum purity) inserted in a thin glass tube.
All used reagents were of analytical grade and used as-received from the suppliers, without further purification. Sulfuric acid (95-97%) was purchased from Merck, and potassium iodide (KI) was purchased from Sigma-Aldrich. Iron (II) sulfate heptahydrate (FeSO4.7H2O) was purchased from AppliChem. Hydroxylammonium chloride (NH2OH.HCl) was an AnalaR Normapur® ACS reagent (BDH). The nitrogen gas was of grade 5.0, with 99.999% minimum purity, and it was supplied from The Jordan Gases Company (Amman, Jordan).
Procedures
Every experiment was started by reproducing the platinum electrode cyclic voltammogram. The cyclic voltammogram reproduction of a polycrystalline platinum electrode with the clear hydrogen-oxygen adsorption/desorption features constitutes a real evidence for the electrochemical system cleanliness, including the solution, electrodes and the purging gas.
The platinum electrode coating process was preceded by immersing a part of it in the supporting electrolyte (0.5 M H2SO4 +10-2 M KI) for five min., at a deposition potential of 0.2 V (double layer region), to make iodine ion adsorb at the platinum electrode surface as a neutral iodine atom. The electrode was extensively in situ rinsed with an iodine-free 0.5 M H2SO4 solution. The cyclic voltammogram for the iodine-coated platinum electrode was recorded between its potential window limits, -0.25 V and 0.85 V. The absence of both hydrogen adsorption-desorption and oxide formation/removal voltammetric features, except for the charging current, is attributed to the complete coating of the platinum electrode surface with iodine.
The water samples were supplied from the water laboratories of the Jordanian ministry of water and irrigation. These samples were collected from three different deep pioneer wells in the south of Jordan: Qatraneh, Swaqah and Qastal. The depth of the pioneer wells is between 1000 to 1100 meters. The collected water samples were directly treated with concentrated nitric acid to keep iron ions in the ferrous state. The samples were kept in tightly closed clean plastic bottles until the time of analysis. The collected samples were analyzed for their iron (II) content, in triplicate, using an ICP-OES instrument.
Anodic stripping voltammetric measurements for iron (II) determination were carried out at the deposition potential of -0.15 V (vs. Ag/AgCl/[Cl-] = 1.0 M), for 240 sec. After 60 sec. of solution equilibration, the electrode was scanned between -0.15 V and 0.85 V, at a scan rate of 50 mv/sec.
Once the water samples were started to be analyzed, few crystals of hydroxylamine were added to prevent any iron (II) to iron (III) ions oxidation. Each of the samples was analyzed three times.
Results and discussion
Method results and discussion
A representative reproducible cyclic voltammogram for the clean platinum electrode surface, along with a typical anodic stripping voltammogram of the iodine-coated platinum electrode, are shown in Fig. 1. The modified iodine-coated platinum electrode linear sweep voltammogram demonstrates a complete absence of the hydrogen and oxygen adsorption/desorption features of the platinum electrode surface cyclic voltammogram. The iodine-coated platinum electrode surface is stable at a potential range from -0.25 V to +0.85V.
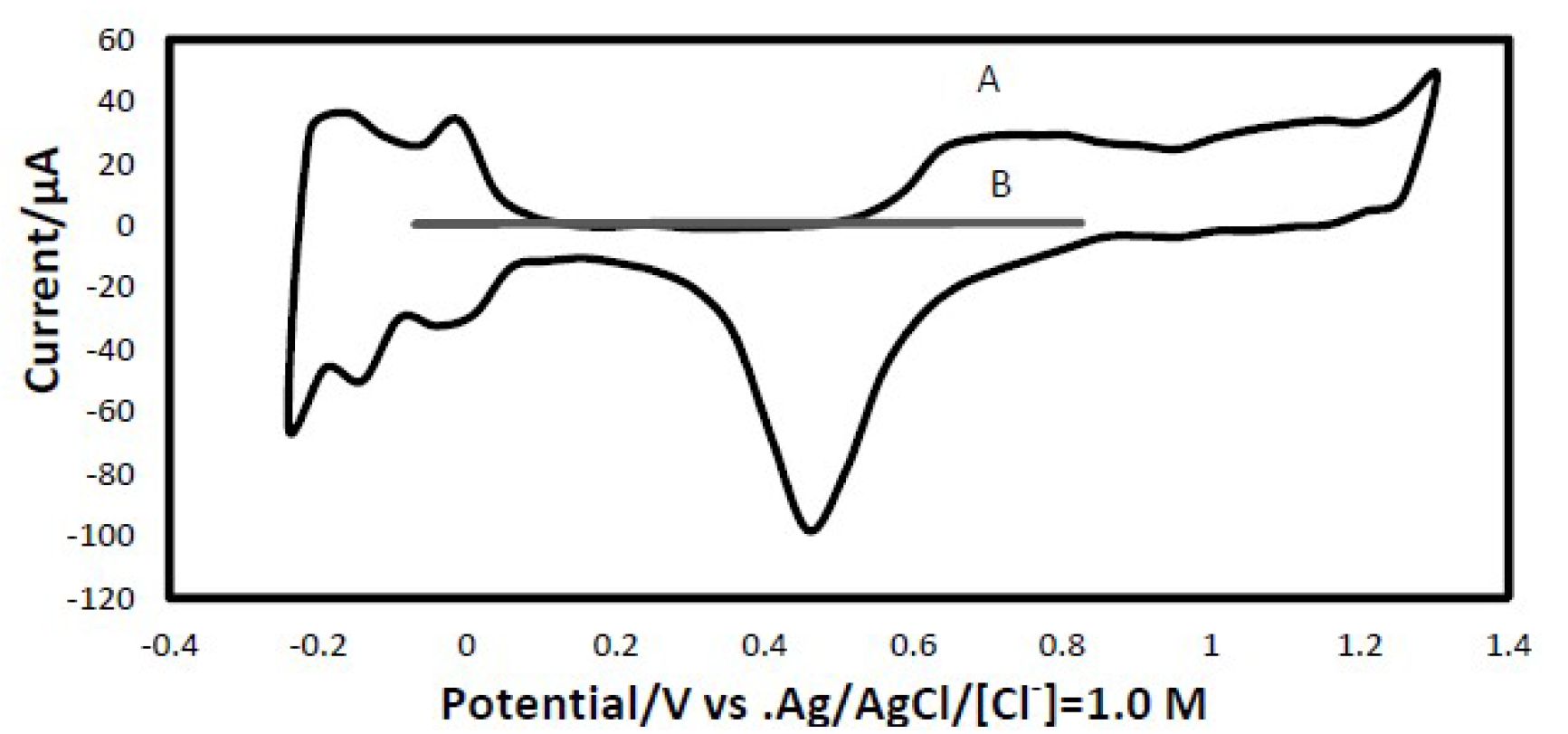
Figure 1 (A) Cyclic voltammogram of the polycrystalline platinum electrode. (B) Anodic stripping voltammogram of the iodine-coated platinum electrode. Both i-E scans were recorded in iodine-free 0.5 M H2SO4 . Scan rate = 50 mV/s.
Fig. 2 shows a set of anodic stripping voltammograms for 0.5 M H2SO4 solutions, with a series of iron (II) standard solutions concentrations ranging from 2 to 10 ppm. Under the optimized conditions, the iron oxidation peak current centered ca. 0.72 V.
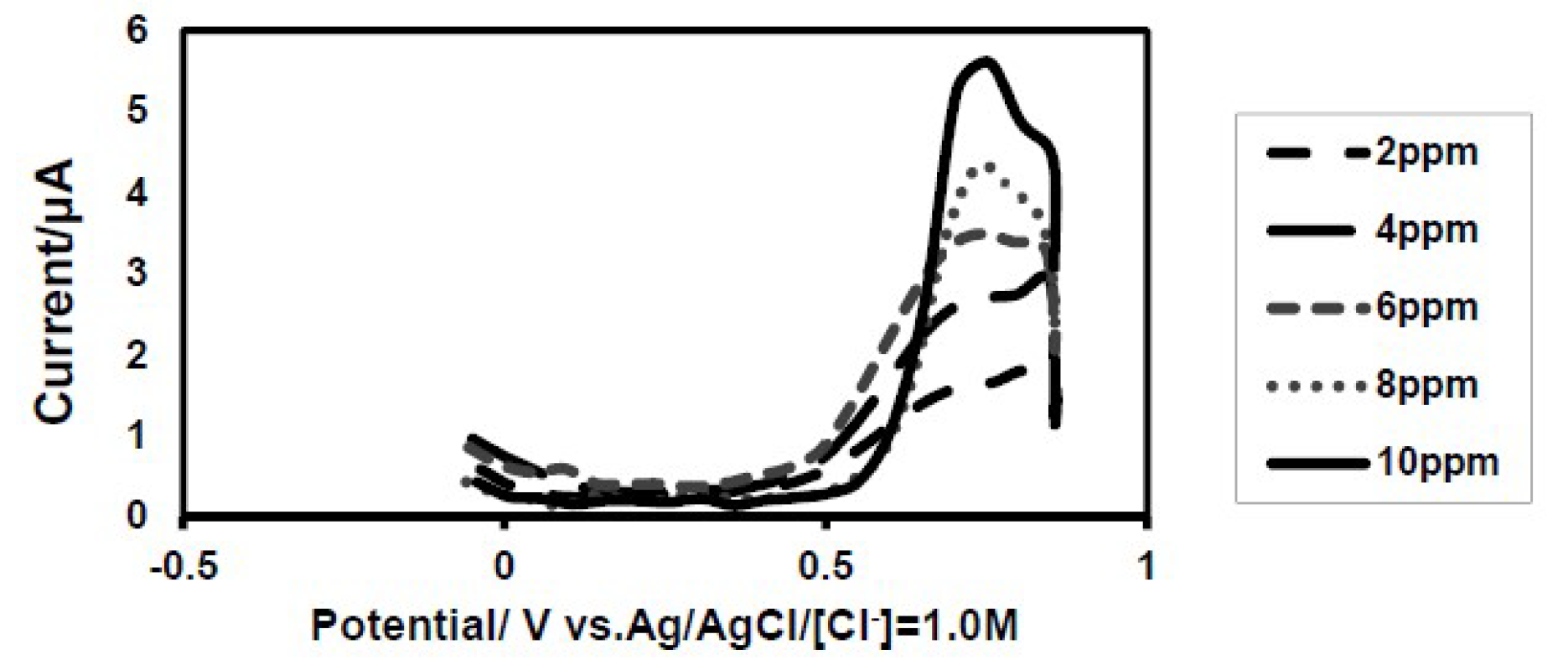
Figure 2 Anodic stripping voltammograms of an iodine-coated electrode recorded in a 0.5 M H2SO4 solution containing (−−) 2 ppm Fe (II), (― ―) 4 ppm Fe (II), (---) 6 ppm Fe (II) (….) 8 ppm Fe (II) and (―)10 ppm Fe (II). Scan rate = 50 mv/sec.
The calibration curve was constructed by plotting the anodic peak currents extracted from the linear seep voltammograms against the Fe (II) concentration in the solution. The linearity and the linear range were obtained by triplicate analysis of the extended concentration levels, from 1 to 100 ppm, as shown in Fig. 3.
The calibration curve shows excellent linearity with a linear regression value of R2 = 0.996 and the calibration equation (Eq.1) is:
i (μA) =0.511CFe(II)+0.057 (1)
where i is the value of anodic peak current attributed to Fe to Fe (II) oxidation, after the preconcentration period, and C is the Fe (II) standard solutions concentration. The calibration curve extension of indicates no deviation from the linearity, as shown in Fig. 3.
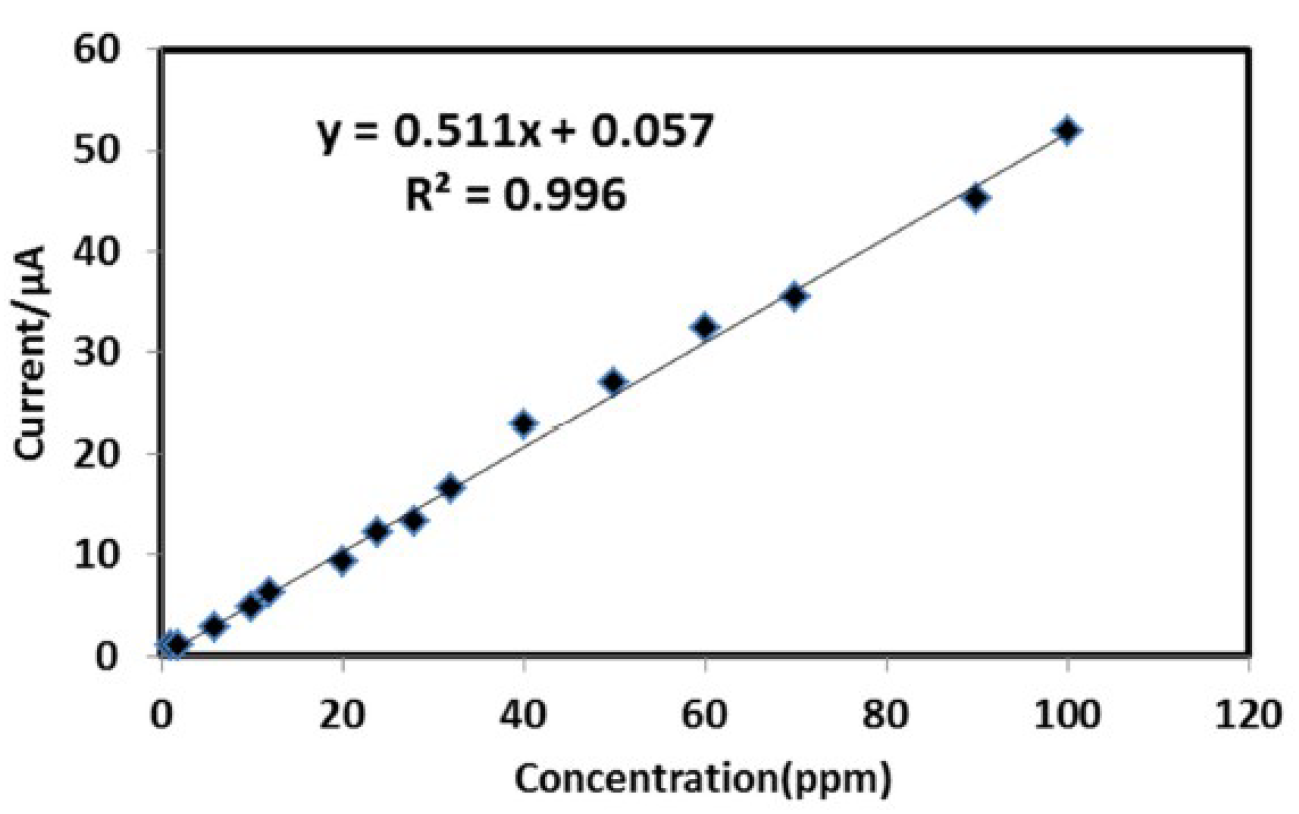
Figure 3 Calibration curve showing the correlation between iron (II) concentration and the anodic peak current obtained from iron (II) voltammogram in 0.5 M H2SO4, at an iodine coated electrode. Scan rate = 50 mv/s.
The precision of the developed method was evaluated by analyzing the aliquot of a 6 ppm standard solution for ten times. The standard deviation and variation coefficient were 4.23% and 1.28%, respectively.
The lowest limit of detection (LOD) estimation based on S/N = 3 and the limit of detection (LOQ) based on S/N = 10, for the developed analytical method, were 0.26 ppm and 0.85 ppm, respectively.
Potential interferences
Heavy metals, such as Zinc, chromium, cadmium, manganese, copper, cobalt, nickel, molybdenum, silver, mercury, arsenic, bismuth, antimony and lead are common in ground water [29].
The proposed anodic stripping analysis method using an iodine-coated platinum electrode showed a voltammetric response towards copper, silver, bismuth, mercury and antimony, while no response was recorded towards the remaining metal ions (Fig. 4).
Mercury is undetected in ground water, while antimony is found in groundwater in very low concentrations [30]. The silver and bismuth oxidation peaks (Fig. 4) do not interfere with the iron oxidation peak.
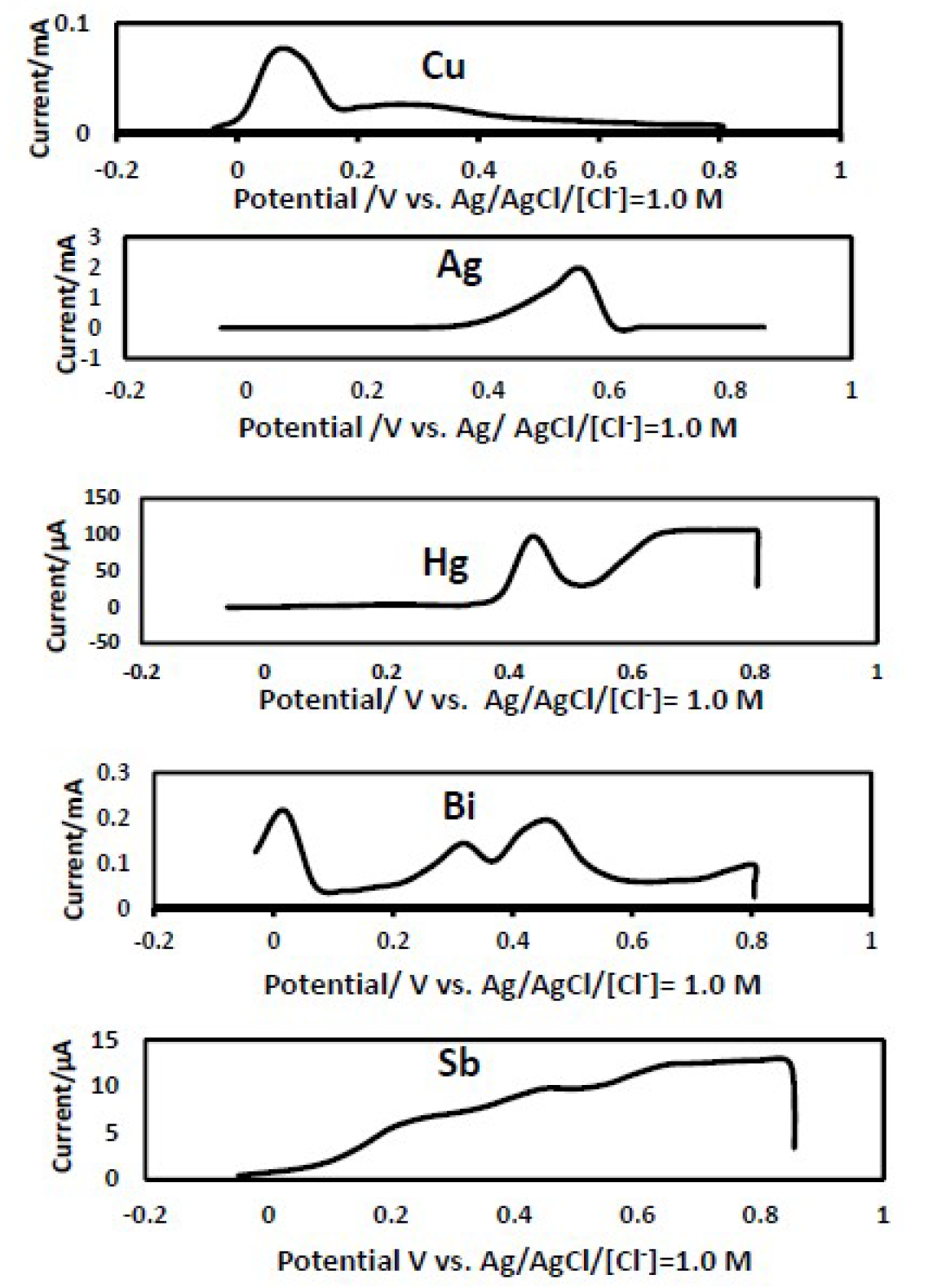
Figure 4 Current responses recorded after the injection of 4 ppm of Cu, Ag, Hg, Bi and Sb ions in an experiment performed with an iodine-coated platinum electrode in a 0.5 M H2SO4 supporting electrolyte. Scan rate = 50 mv/sec.
Analysis of ground water samples
The water samples were analyzed for their iron (II) content using the standard addition method. Table 1 displays the results obtained for iron (II) determination in the tested three water samples analyzed using the anodic stripping method at an iodine-coated electrode.
Table 1 Analysis of deep groundwater samples for iron (II) by the developed anodic striping method at an iodine-coated platinum electrode.
| Sample no. | Water sample | Nominal conc (in ppm) of water sample as analyzed by ICP-OES instrument* | Average determined conc (in ppm) | Standard deviation | 95% confidence limits | 95% confidence interval | Relative error | Coef of variat (n=3) |
|---|---|---|---|---|---|---|---|---|
| 1 | Qatraneh | 6.63 | 6.38 | ±0.42 | 6.38±1.044 | 5.336-7.468 | -3.77% | 6.58% |
| 2 | Swaqah | 6.32 | 6.29 | ±0.32 | 6.29±0.795 | 5.495-7.085 | -0.48% | 5.09% |
| 3 | Qastal | 4.04 | 4.14 | ±0.15 | 4.14±0.373 | 3.767-4.513 | +2.48% | 3.62% |
*These values are according to the analysis results by water laboratories, ministry of water and irrigation, Jordan.
The estimation of data in Table 1 shows that all nominal values are within the 95% confidence interval, which means the absence of determinate errors in the measurements. The relative error values of the three analyzed water samples were all below 5%, which attests to the accuracy of the developed method. The coefficient of variation values are between 3.62% and 6.58%, which provides an obvious evidence for the precision of the developed analytical method. Thus, as it is presented in Table 1, there is a satisfactory agreement between the results achieved by the optimized anodic stripping method and the reported values by the ICP-OES instrument for the analysis of the same water samples. The paired t-test was applied to examine the significant difference between the values measured by the developed anodic stripping method and the values obtained by the ICP-OES analysis technique. Based on the t-test evaluation, the measured t value is -0.041, while the critical t-value is 4.30, at p = 0.05 [31]. The degree of probability is lower than the conventional level of 5%. This confirms the truth of the null hypothesis, and indicates that there is no difference between the means of the results obtained by the application of the developed voltammetric method and the ICP-OES method. This result attests to the reliability of its application for iron determination in water samples.
Table 2 Analytical parameters of the developed method compared to some common methods for iron analysis in water samples.
| Reference | Matrix | LOQ | LOD | Linear range | Method |
|---|---|---|---|---|---|
| [32] | Surface water | 0.0024 mg/L | 0.0071 mg/L | 0.005-0.2 mg/L | Inductive coupled plasma-optical emission spectroscopy |
| [33] | Aqueous solutions | - | 0.5 µg/L | 0.001-100 µg/L | Spectrophotometry |
| [34] | Drinking water | 7.1 µg/L | 2.17 µg/L | Atomic absorption spectroscopy | |
| This work | Deep ground water | 0.85 mg/L | 0.26 mg/L | 1-100 mg/L | Anodic striping voltammetry |
The developed green analytical method has the advantages of simplicity in preparation procedure, cost and time-saving. A comparison between some common analytical methods and our developed method for iron analysis is given in Table 2.
Conclusions
In the present work, the use of an optimized anodic stripping method, based on the simply prepared iodine-coated platinum electrode for iron determination in water, was demonstrated. The modified iodine-coated platinum electrode is stable and fit for one day analysis work, unless the electrode potential is scanned beyond the potential window limits, or there is a sudden over current or overpotential.
The linearity was obtained for concentrations ranging from 1 to 100 ppm, with a detection limit of 0.26 ppm. The absence of any possible interference from other metal ions in the water matrix supports the applicability of this method, with some exception for both antimony and mercury metal ions which are found in very low concentrations in deep groundwater resources. The analysis of water samples and the statistical analysis of the results showed the absence of determinate errors, and confirmed the null theory; there was no significant difference between the results and the reported values by the water laboratories of the Ministry of water and irrigation. The advantages of the proposed method include the simplicity of preparation and reproduction of the iodine-coated platinum electrode. Additionally, the green and inexpensive instrumentations recommend this method for trace iron electroanalysis.

