











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
The use of inhibitors is one of the best options for metals protection against corrosion (1, 2). However, the process mechanistic is very complex, hence, the usefulness of computational tools to elucidate the interaction between the adsorbed inhibitor molecules and the corroding metal surface.
Most corrosion inhibitors, which slow down the corrosion rate of a metal through the mechanism of adsorption onto the metal surface , (3) are organic compounds containing hetero atoms (such as N, O, S and P), as well as the π orbital character of donating electrons into their carbon chain systems.
Heterocyclic systems rich in electrons, such as phenyls or thiadiazoles, have constantly attracted the attention of researchers in the corrosion field. (4, 5) The series of 2,5-bis (n-pyridyl) -1,3,4-thiadiazoles (n-PTH: n = 2-4), 2,5-bis (phenyl) -1,3,4-thiadiazole (DPTH), 2,5-bis (4-methoxyphenyl) -1,3 molecules, 4-thiadiazole (4-MTH), 2,5-bis (4-dimethylaminophenyl) -1,3,4-thiadiazole (4-DATH), 2,5-bis (4-methylphenyl) -1,3,4- thiadiazole (4-MPTH), 2,5-bis (4-nitrophenyl) -1,3,4-thiadiazole (4-NPTH) and 2,5-bis (4-chlorophenyl) -1,3,4-thiadiazole (4-CPTH), are known for their corrosion inhibiting properties (6). The Electrochemical Impedance Spectroscopy (EIS) measurements of n-PTH (n = 2 - 4) corrosion inhibition on mild steel, in different acidic media (1 M HCl, 0.5 M H2SO4 and 1 M HClO4), have detected a noticeable inhibition, which was assigned, through primary quantum calculations, to the electron density of heterocyclic rings N and thiadiazole ring S atoms (7) In the same way, the inhibition performance, on mild steel in 1 M HCl solutions, of DPTH, 4- MTH, 4- DATH, 4-MPTH, 4-NPTH and 4-CPTH, has been theoretically investigated by using a limited number of quantum molecular descriptors, such as dipole moment (μ), highest occupied (EHOMO) and lowest unoccupied (ELUMO) molecular orbitals and the differences between them (HOMO-LUMO gap) . (8) Similarly, the macrocyclic polyethers, such the series of macrocyclic polyether compounds (n-MCTH, n: 1-5), which are shown in Fig. 1, synthesized by Bentiss et al. (9), also exhibit an electronic richness, since they contain one thiadiazole ring, polyether macrocyclic oxygen and two phenyls rings. Their corrosion inhibitory effect on carbon steel in 1 M HCl has been reported earlier by the Weight Loss Method (WLM), EIS and potentiodynamic polarization (10, 11, 12). It was shown that the macrocyclic polyethers inhibition efficiency increased with their concentration in the order of 5-MCTH > 4-MCTH > 3-MCTH > 2-MCTH > 1-MCTH. The same authors tried to predict the experimental inhibition efficiency classification via B3LYP/6-31G (d, p) DFT level. Based only on a limited number of global descriptors (EHOMO, ELUMO, ΔE, μ, molecular area and CCCS dihedral angle), the prediction remains very incomplete towards a deep understanding of the anti-corrosive adsorption. That is why we have largely explored the ab initio and molecular dynamics interaction of the iron surface with n-MCTH, to elucidate the impact of the thiadiazole nucleus enriched by the aromatic rings electrons, and especially that of the oxygen atoms of the macrocycle polyether part.
Recently (13), we have tried to elucidate the mechanism of two macrocyclic polyether compounds, 1-MCTH and 2-MCTH, and their corrosion inhibition protonated forms, as well as the intrinsic reasons for their inhibition efficiency classification, by using DFT and Dynamic Monte Carlo calculations. Therefore, as a logical continuation of this work, we present ab initio and molecular dynamics investigations of iron surface interactions with the remaining molecules of the n-MCTH (n = 1-5) series, which are 3- MCTH, 4-MCTH and 5 MCTH.
Calculation details
Reactivity quantum global and local descriptors
All geometry optimizations and quantum chemical calculations were performed using B3LYP/6-31G (d,p) level (14), by means of the Gaussian 2009 (15) and Gaussview 05 package. To model the solvation effect, the PCM (Polarizable Continuum Model), considering the solvent as a continuum and the solute in its cavities, was adopted . (16)
As well described in our earlier work (13), quantum electronic parameters investigation concerns:
The molecular descriptors such as the energy of highest occupied molecular orbital (EHOMO), the energy of the lowest unoccupied molecular orbital (ELUMO), the energy gap (∆E) (17), the global hardness (η) (18), the absolute electronegativity (χ), the overall softness (S), the electrophile index (ω), the fraction of electrons transferred (ΔN) (19) and the total exchange energy (ΔE) back-donation.
The local descriptors, such as molecular electrostatic potential (MEP), NBO charges, NBO energy of hyper-conjugative interactions (17) and Fukui indexes (fk-, fk+ and fk0) of the nucleophilic, electrophilic and radical attack, respectively (20) (19), knowing that fk- higher value gives rise to a nucleophilic site, and that fk+ higher value gives rise to an electrophilic site. The formulations of these electronic characteristics were mentioned elsewhere (21, 22).
Dynamic Monte Carlo simulation
Likewise for n-MCTH (n:1-2) (13), the Metropolis Dynamic Monte Carlo simulation, using the adsorption locator and Forcite codes implemented in the Material Studio 7.0 software (23), was carried out to locate the stronger adsorption between the inhibitor molecules and the iron surface, i.e. the higher negative interaction energy value. Although Fe (110) plane is a densely-packed surface, the crystallographic Fe (111) surface was chosen because of the lower value of its formation energy (24). The structures of all components of the inhibitor-iron surface system were optimized with COMPASS II force field, and the simulation box (17.89×17.89×38.34Å) was used with periodic boundary conditions, in order to simulate a representative part of an interface. The Fe (111) plane was next enlarged to a (9 × 9) super cell. After that, a vacuum slab with 3.0 nm thickness was built above the Fe (111) plane. To mimic the real experimental corrosion environment, 100 molecules of water were added to the simulation box.
Results and discussion
In addition to the solvent effect, the structural properties, mainly, the level of planarity of n-MCTH (n = 3-5) molecules, and their competitive protonated forms, on the one hand, and their electronic properties, on the other hand, have an impact on their inhibition efficiency.
Regio-selectivity of neutral species isolated and in an aqueous solution
The optimized geometries of 3-MCTH, 4-MCTH and 5-MCTH are illustrated in Fig. 2.
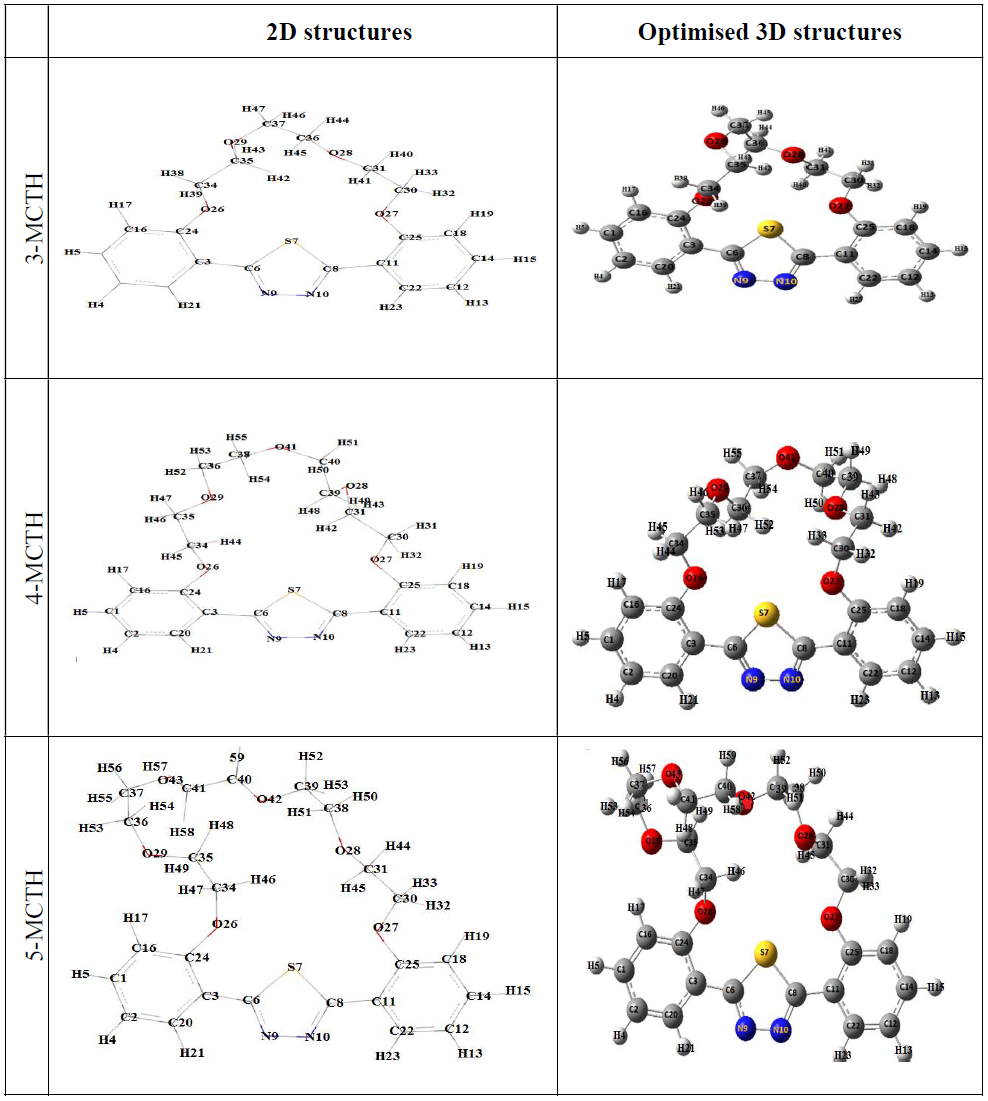
Figure 2 Schematic representation of 2D structures and 3-MCTH, 4-MCTH and 5-MCTH optimized structures, at B3LYP / 6-31G **.
Selected geometrical parameters
As confirmed by a structural comparison of 1-MCTH and 2-MCTH, the increase in the number of oxygen atoms in 3-MCTH, 4-MCTH and 5-MCTH makes the ring containing oxygen less constrained. Indeed, the distance between the two aryls increases (C24-C25 distance increases up to 0.15 A° from 3-MCTH to 4-MCTH and 5-MCTH), and the dihedral angle between the two aryls described by C20-C3-C11-C22 decreases, until making them quasi-coplanar for 5-MCTH (Tables 1 and 2). It is the same with the thiadiazole ring that also becomes coplanar with aryls, as C24-C3-C6-S7 and C25-C11-C8-S7 have reached almost zero. However, the aqueous solvent acts against the planarity of these molecules, as C20-C3-C11-C22 and C25-C11-C8-S7 increase up to 20° for 5-MCTH, e.g.
Table 1. Bond distance (Å) for the neutral inhibitors 3-MCTH, 4-MCTH and 5-MCTH optimized structures and their double protonated forms (N9H+N10H+), in vacuum (G) and in an aqueous solution (A), at B3LYP/6-31G**.
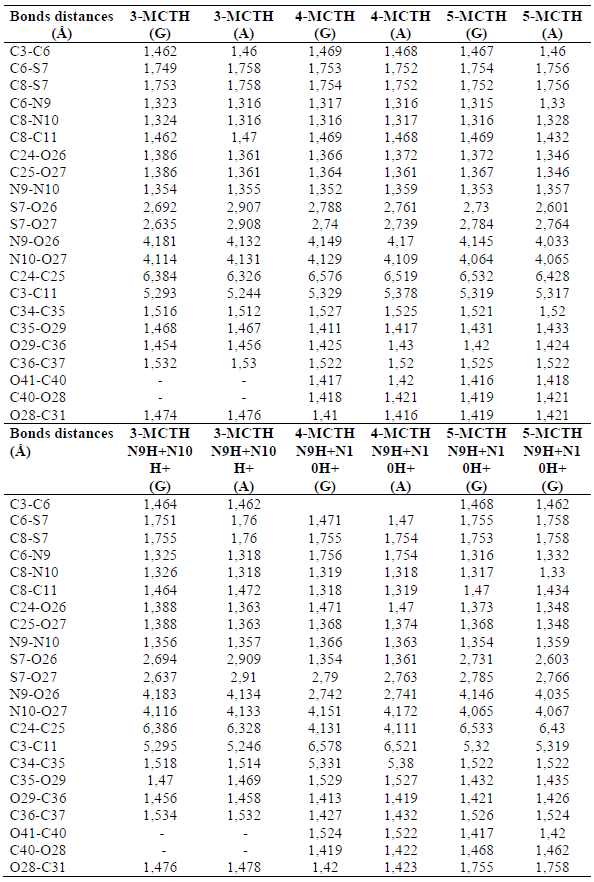
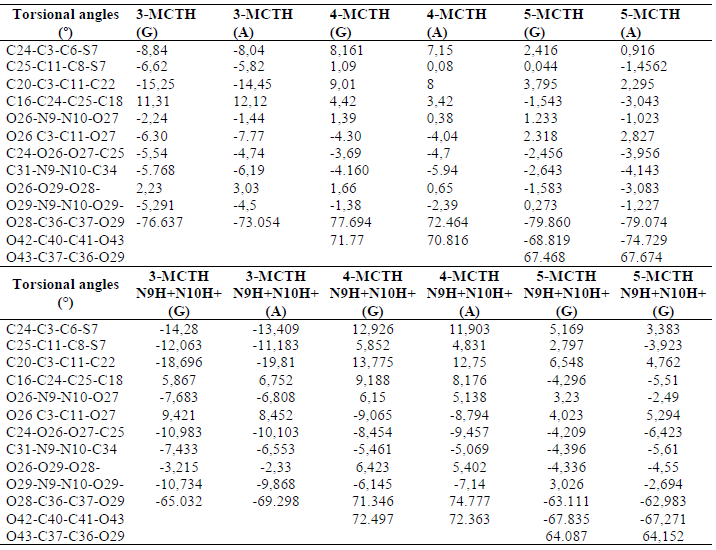
Table 2 Torsional angle (º) for the 3-MCTH, 4-MCTH and 5-MCTH neutral inhibitors optimized structures and their double protonated forms (N9H+N10H+), in vacuum (G) and in an aqueous solution (A), at B3LYP/6-31G**.
Taking into account the structural aspect, 5-MCTH can be considered the most appropriate corrosion inhibitor, because it has a very planar first molecular part, and a second part with a large recovering surface. In comparison with the available experimental data, 4-MCTH X-ray crystallographic data show that its structure belongs to the C 2/c space group of the monoclinic system, and that it is practically plane. Moreover, the phenyl rings are shifted relative to the thiadiazole ring by an angle equal to 4.93°. In addition, a weak hydrogen bond connects the polyether macrocyclic oxygen and phenyls, providing two-dimensional stability of the crystalline system (10).
Molecular quantum chemical parameters
The results presented in Fig. 3 show that HOMO is delocalized throughout all aryl and thiadiazole rings, except the S atom of thiadiazole, and C16, C18, C20 and C22 of aryl rings, which are excluded from the electron donation regions. LUMO is spread out on all the aryl and thiadiazole rings, providing them high tendency to accept electrons from nucleophilic species. However, the formation of the LUMO orbital does not involve the Pz orbitals participation. Moreover, oxygen atoms are less concerned by the electrons transfer effect.
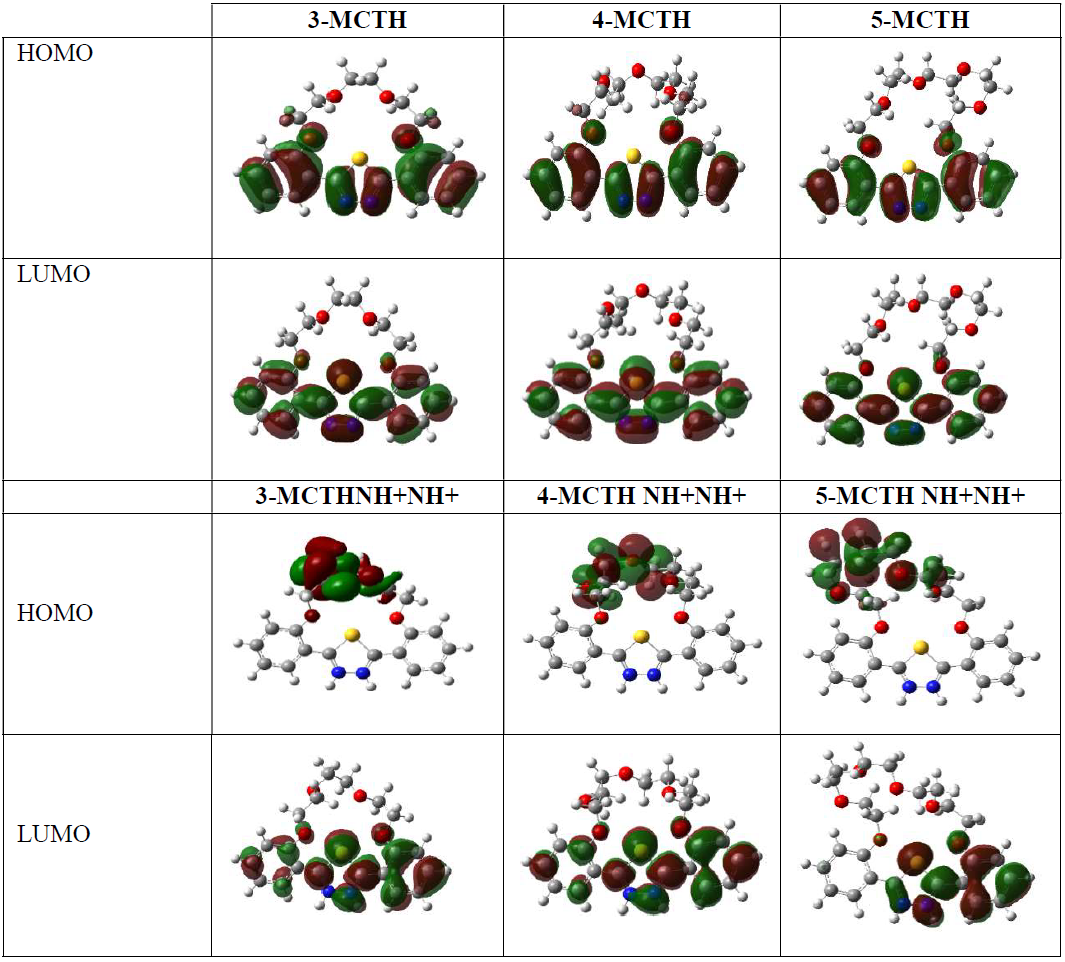
Figure 3 HOMO and LUMO 3-MCTH, 4-MCTH and 5-MCTH neutral inhibitors and their double protonated forms (N9H+N10H+), in vacuum, at B3LYP/6-31G**.
Molecular quantum parameters allow us to have more information about the global reactivity of inhibitors and their potential to interact with the metal. From Table 3, we notice that 5-MCTH has the highest EHOMO, indicating its ability to easily give electrons to electrophilic sites, and its higher reactivity with the lower ∆E gap, as ELUMO remains practically constant. The molecular reactivity could also be expressed by the global softness values and the fraction of electrons (∆N) transferred from the inhibitor to iron, knowing that the latter would preferentially interact with inhibitors having high S and low ƞ (ƞ= ∆E /2) values.
Table 3 Some quantum chemical parameters for the 3-MCTH, 4-MCTH and 5-MCTH neutral inhibitors, in vacuum (G) and in an aqueous solution (A), at B3LYP/6-31G**.
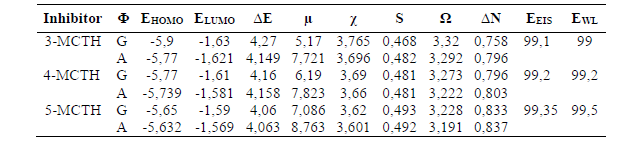
With higher S and ∆N, 5-MCTH is more reactive than the other inhibitors, which is confirmed by its lower electronegativity and larger dipole moment that inform about the molecule polarity (25). This decrease in electronegativity led to a reduction in the global electrophilicity index, and, therefore, to an increase in the nucleophylic character, as the nucleophile is characterized by a lower ω value.
All energy values are in ev, the dipole moment (μ) is in Debye, S is the global softness in ev-1, and EEIS and EWL are the average experimental percentage inhibition efficiency (%), obtained, respectively, from electrochemical impedance spectroscopy and weight loss method, as given in (10,12).
As expected, the solvent addition substantially increases the molecules dipole moment, as their polarity changes seriously. It also reduces significantly the electrophilicity index (ω), while only a slight fluctuation has reached the other parameters, without any change in the reactivity classification order.
Combining these results with those of the frontier orbital picture analysis for molecules isolated and in the aqueous solution, we can conclude that 5-MCTH is more reactive in both gaseous and aqueous phases.
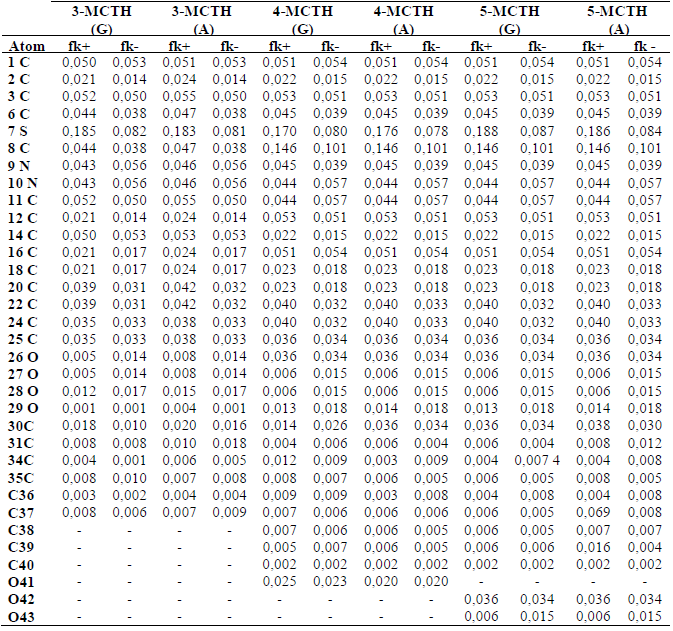
Table 4 Condensed Fukui functions on the 3-MCTH, 4-MCTH and 5-MCTH neutral inhibitors atoms, in vacuum (G) and in an aqueous solution (A), at B3LYP/6-31G**.
Local reactivity and population analyses
Local reactivity based on FUKUI indices
It can be seen from Table 4, for the three molecules, that the atoms which are the most susceptible sites for the nucleophilic or electrophilic attacks belong to the thiadiazole and aryls rings, as they present fk+ and fk- high values, such as C1, C3, C6, C8, C11, N9, N10, C11 and C14.
Molecular Electrostatic Potential
The MEP surface picture of the three molecules, given in Fig. 4, reveals that the region of high negative charges (sites rich in electrons, represented by the red colour) is seen around the N9 and N10 nitrogen atoms that are susceptible to an electrophilic attack.

Figure 4 Molecular electrostatic potential contour map (MPE) for the 3-MCTH, 4-MCTH and 5-MCTH neutral inhibitors optimized structures and their double protonated N9H+N10H+ optimized forms, in vacuum (G), at B3LYP/6-31G**. (Red: strong negative electrostatic potential (EP); yellow: moderately negative EP; blue: strong positive EP; green: moderately positive EP).
The protonation process of these two sites confirms this, since it weakens their electronic abundance, making them strongly positive, with high electrostatic potential energy value, represented by the blue colour. Moreover, the MEP surface also shows a substantial distribution of negative charge in the central oxygen atom, compared to the peripheral oxygen atom for 4-MCTH, as found for 2-MCTH in previous work, i.e. when the oxygen atoms number is odd(13) .
It should be noted, though, that C8 fk+ and fk- became more important for 4-MCTH and 5-MCTH, compared to those of 3-MCTH (about 0.15, instead of 0.04), and that C14 fk+ and fk- decreased remarkably from 0.05 (3-MCTH) to 0.02 (4-MCTH and 5-MCTH), while their values increased from 0.02 to 0.05, for C12 and C16. On the other hand, the sulphur site has important fk+ values, of the order of 0.18, for 4-MCTH and 5-MCTH, and of 0.17, for 3-MCTH, which implies that sulphur is more susceptible to receive electrons than to donate them to the metal surface. Concerning the solvent effect on these indices, we noted that water, as solvent, had no significant effect on the local reactivity.
As it can be seen, the results of the local reactivity based on Fukui indices corroborate those obtained by the HOMO and LUMO analysis.
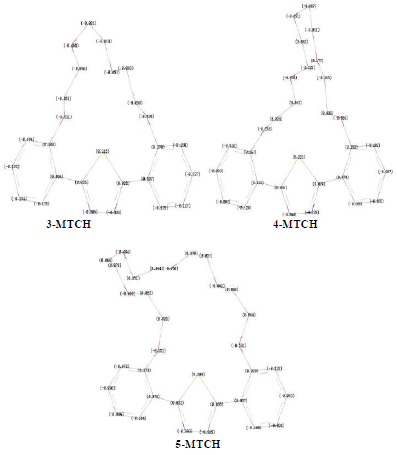
Figure 5 NBO charges of the optimized neutral inhibitors 3-MCTH, 4-MCTH and 5-MCTH in vacuum, at B3LYP/6-31G**.
Natural population atomic charges and natural bond orbital analyses
The distribution of the molecular charge in terms of NPA (Natural Charge Population Analysis) is shown in Fig. 5. The partial atomic charges of nitrogen take a value of the order of -0.3, while those of oxygen are comparable to -0.5, for the three inhibitors .
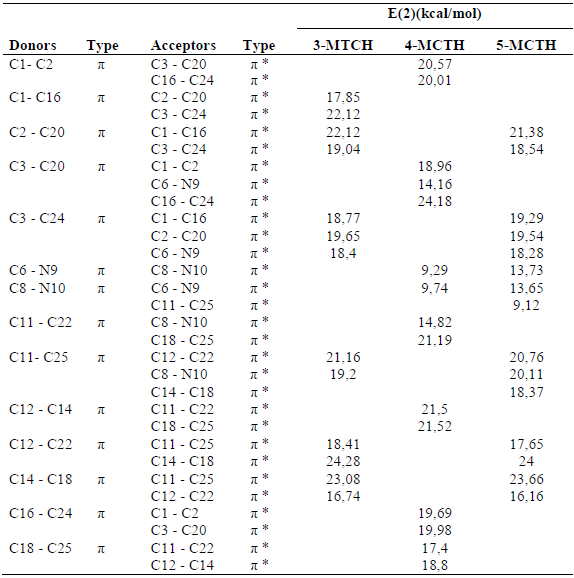
With regard to the NBO analyzes, Table 5 shows that the strongest intramolecular charge transfer concerns the πCC - π*CC transition within the same phenyl radical, with a stabilization energy of approximately 24 kcal/mol, which concerns πC12C22- π*C14C18, for 3-MCTH and 5-MCTH, and πC3C20 - π*C16C24, for 4-MCTH.
Table 5 Second Order Perturbation theory analysis of Fock matrix in NBO basis for 3-MCTH, 4-MTCH and 5- MCTH, at B3LYP/6-31G**. (E(2) means energy of hyper conjugative interactions).
Regioselectivity of isolated protonated species and in an aqueous solution
By comparing the proton affinity (PA) at the different possible sites for 1-MCTH and 2-MCTH, we have shown that the protonation and double protonation on N9 and N10 nitrogen atoms are preferred to those on O26 and O27 oxygen atoms. Moreover, the aqueous solvation had a stabilizing effect on the protonation process, and preserved the same order of protonation preference. For these reasons, we have considered, for these three molecules, only the double protonation on nitrogen atoms.
The 3-MCTHN9H+N10H+, 4-MCTHN9H+N10H+ and 5-MCTHN9H+N10H+ optimized geometries are shown in Fig. 6. The evaluations of the proton affinity (26) show that the protonation process is exothermic, meaning that all inhibitors have a tendency for protonation. It should be also noted that the proton affinity (PA) became more important for 5-MCTHN9H+N10H+ and 4-MCTHN9H+N10H+.
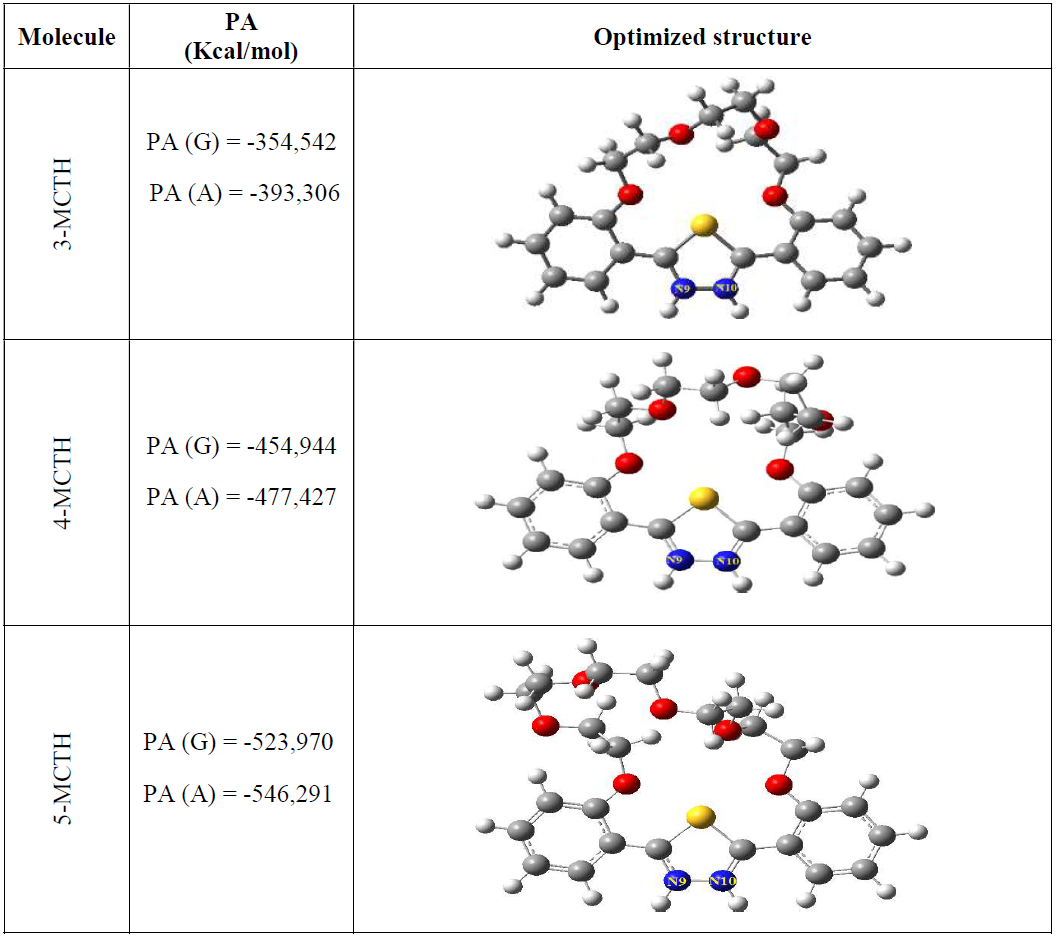
Figure 6 Double protonated N-N species of 3-MCTHN9H+N10H+, 4- MCTHN9H+N10H+ and 5- MCTHN9H+N10H+, in vacuum (G) and in an aqueous solution (A), at B3LYP/6-31G**, and the corresponding proton affinity values (kcal/mol).
Selected geometrical parameters
The geometric parameters selected for the double protonated species are also reported in Tables 1 and 2. The double protonation on N9-N10 resulting in n-MCTHN9H + N10H + (n = 3 - 5) did not show any significant variation in binding lengths, including those sensitive to double protonation in 1-MCTH and 2 -MCTH, such as C3-C6 and C11-C8, separating the aromatic rings from the thiadiazole ring, as well as C6-N9 and C8-N10, within the thiadiazole ring, and the interaomic distance between the two C24-C25 phenyls. In the same context, the double protonation on the two nitrogen atoms had a variation not exceeding 5° on the torsion angles between the two aromatic rings, expressed by C20-C3-C11-C22 or C16-C24-C25-C18, and the twist angle between the thiadiazole ring and the aromatic rings, expressed by C24-C3-C6-S7 or C25-C11-C8-S7.
As a result, and contrary to 1-MCTH and 2-MCTH, n-MCTHN9H + N10H (n = 3 - 5) does not appear to have a degree of planarity very different from its neutral counterparts (Fig. 7). The same observation can be made for the solvation of protonated double nitrogen species, since we have not found any significant effect on the degree of planarity, although it acts on the bond lengths.
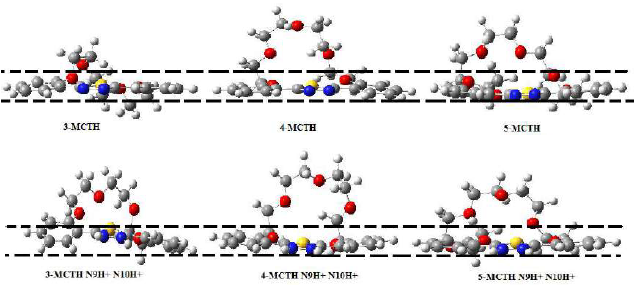
Figure 7 Planarity influencing factor in 3-MCTH, 4-MCTH and 5-MCTH neutral inhibitors and in their double protonated forms, at B3LYP/6-31G**. The region between the dotted lines represents the metal surface.
Quantum chemical parameters of protonated forms isolated and in aqueous solutions
Table 6 reports quantum chemical parameters values of n-MCTHN9H + N10H+ (n = 3 - 5) optimized structures, in gaseous (G) and in aqueous (A) solutions. We note that EHOMO and ELUMO are lower than those of the neutral forms, suggesting that protonation decreases the inhibitor tendency to donate electrons, and increases its tendency to accept electrons, which is confirmed by ΔN negative values, indicating that the electronic exchange is more favorable from the metal towards the protonated inhibitor. With a lower ΔE value and, thus, a higher global softness, S, the protonated species are more reactive than the neutral forms. The electrophilicity character also increases considerably with electronegativity.
With regard to the solvent effect on the protonated species molecular properties, the reactivity expressed by the protonated species ΔE or S is lower than that of the protonated species without solvent. Additionally, the electrophilicity character also decreases drastically, as well as electronegativity. It should also be noted that the ΔN values, which are negative for the isolated protonated forms, become positive under the solvent effect, reflecting the possibility of inversion on the exchange direction, i.e., from the metal to the inhibitor. In addition, the protonation reduces the HOMO orbital space, essentially on the aryl and thiadiazole ring side, thus reducing the cation ability to give electrons, while the same zone remains enriched by electrons in LUMO (Fig. 3).
We can conclude, in the light of the quantum parameters obtained for the n-MCTH (n = 3 - 5) protonated forms, that these are more reactive, in terms of electron acceptance, than the neutral forms; however, the solvent moderates their reactivity. This can be confirmed by the HOMO and LUMO orbitals shape.
Table 6 Some quantum chemical parameters for the 3-MCTH, 4-MCTH and 5-MCTH optimized protonated inhibitors, in vacuum (G) and in an aqueous solution (A), at B3LYP/6-31G**.
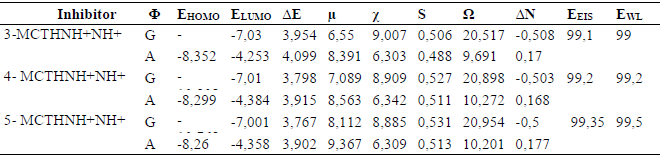
All energy values are in ev, the dipole moment (μ) is in Debye, S is the global softness in ev-1, and EEIS and EWL are the average experimental percentage inhibition efficiency (%), obtained, respectively, from electrochemical impedance spectroscopy and weight loss method, as given in (10, 12).
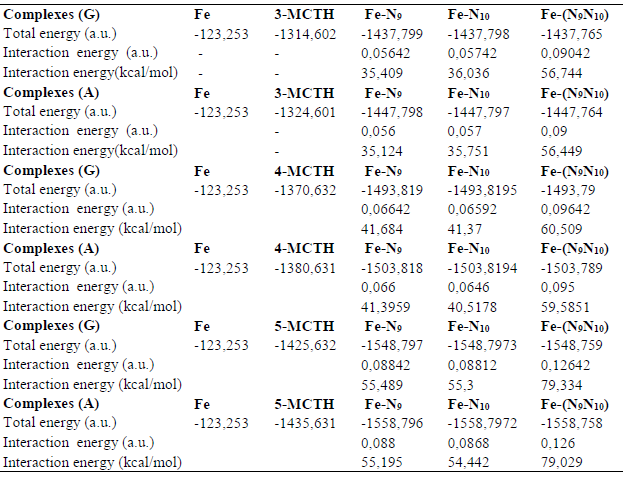
Table 7 B3LYP/6-31G** interaction energies of the 3-MCTH, 4- MCTH and 5-MCTH neutral inhibitors optimized structures, in vacuum (G) and in an aqueous solution (A), with the iron surface.
Metal-inhibitor interaction mechanism in vacuum and with the solvent effect
After having studied the competitiveness between active sites within the same inhibitor, the competitiveness between neutral and protonated forms, and the solvent effect, we were able to deepen our understanding of the inhibition mechanism through the DFT and dynamic Monte Carlo (MC) study of complexation between corrosion inhibitor and iron surface (27).
Table 8 B3LYP/6-31G** interaction energies of the neutral inhibitors 3-MCTH, 4- MCTH double nitrogen protonated forms, in vacuum (G) and in an aqueous solution (A), with the iron surface.
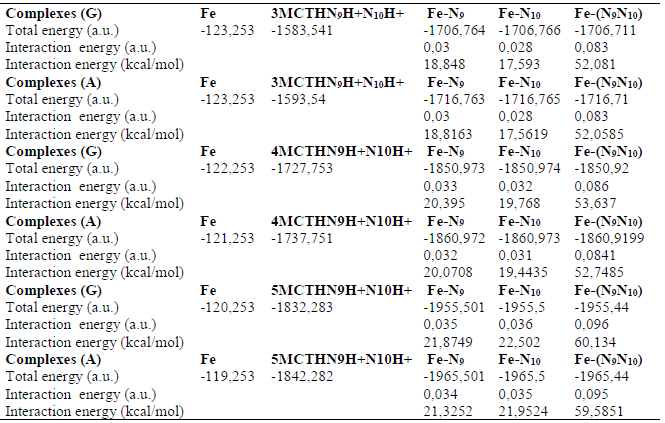
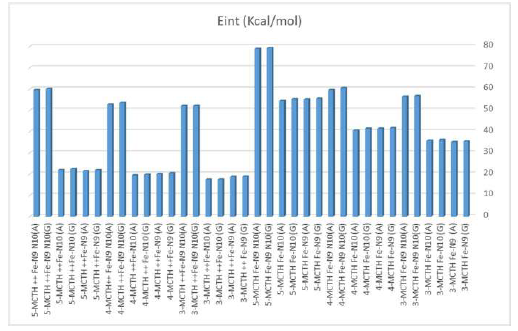
Figure 8 B3LYP/6-31G** interaction energies of the 3-MCTH, 4-MCTH and 5-MCTH neutral inhibitors optimized structures and the optimized double nitrogen protonated forms, in vacuum (G) and in an aqueous solution (A).
Complexation DFT calculation
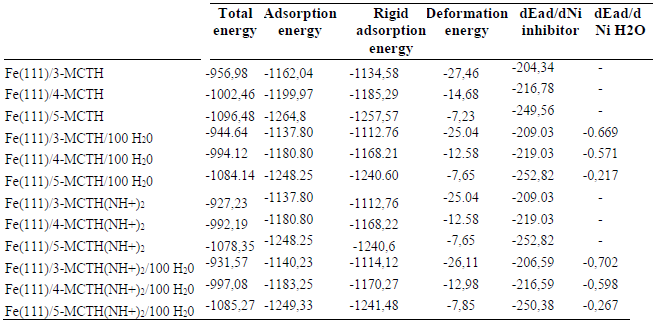
In this part, we evaluated the interaction energy of neutral forms, and of their double nitrogen protonated forms, with the iron metal reduced to a single Fe atom. From the results collected in Tables 7 and 8 and illustrated as a histogram in Fig. 8, we noted, for each molecule, the importance of the interaction involving two nitrogen atoms, compared to that involving one of them, and the importance of the interaction involving a neutral form compared to that of a protonated form. Moreover, we noted the supremacy of the interaction of 5-MCTH neutral and protonated variants over that of their 3-MCTH and 4-MCTH counterparts, which in turn are superior to the interaction of 2-MCTH and 1-MCTH (13).
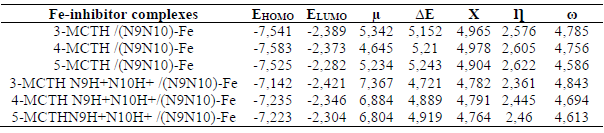
The comparison of the molecular quantum descriptors of the formed complexes is an additional confirmation (Table 9). Indeed, the complexes ΔE and hardness (ƞ) increase, and their electronegativity (χ) and electrophilicity (Ω) decrease, from 3-MCTH / (N9 N10) -Fe to 5-MCTH / (N9 N10) -Fe, confirming the stability of the latter. Table 8 results confirmed also the strength of the complexation due to neutral forms, compared to that due to protonated ones.
We also found that the lengths of the N-Fe bonds decreased with the increase in interaction force, particularly when the Fe atom was simultaneously bonded to N9 and N10. Indeed, Fig. 9 shows this decrease, ranging from 1.953A° to 1.897A° (taken on the average of the two NH lengths), in 3-MCTH and 5-MCTH, respectively, for the neutral forms.
Fig. 9 also shows HOMO, LUMO and total electron density for Fe inhibitor complexes. It confirms the stabilizing effect of the Fe-(NN) interaction in the complex, compared to the Fe-N interaction. However, the supremacy of the interaction of 5-MCTH, compared to the two others, remains hardly perceptible in a visual way, if we exclude the slight reduction of the molecular orbital HOMO or LUMO, from 3-MCTH to 5-MCTH.
Table 9 Some optimized quantum chemical parameters for the Fe-inhibitor complexes and Fe-protonated inhibitor complexes, in vacuum (G).
Dynamic Monte Carlo study of complexation
We point out first that the Dynamic Monte Carlo was performed to look for the lowest configuration adsorption energy of the inhibitor molecules interactions with the iron surface. It provides some vital parameters, such as total surface energy (Etotal), which is the summation of all the adsorbate components energies (28)(29). It also furnishes the inhibitor adsorption energy (Eads) and dEad/dNi energy required when one mole of it is adsorbed onto the metallic surface (30). This informs about the interaction strength between the inhibitor and the metallic surface, knowing that an inhibitor with more negative Eads or dEad/dNi values acts as better adsorbate onto the Fe(111) surface (31).
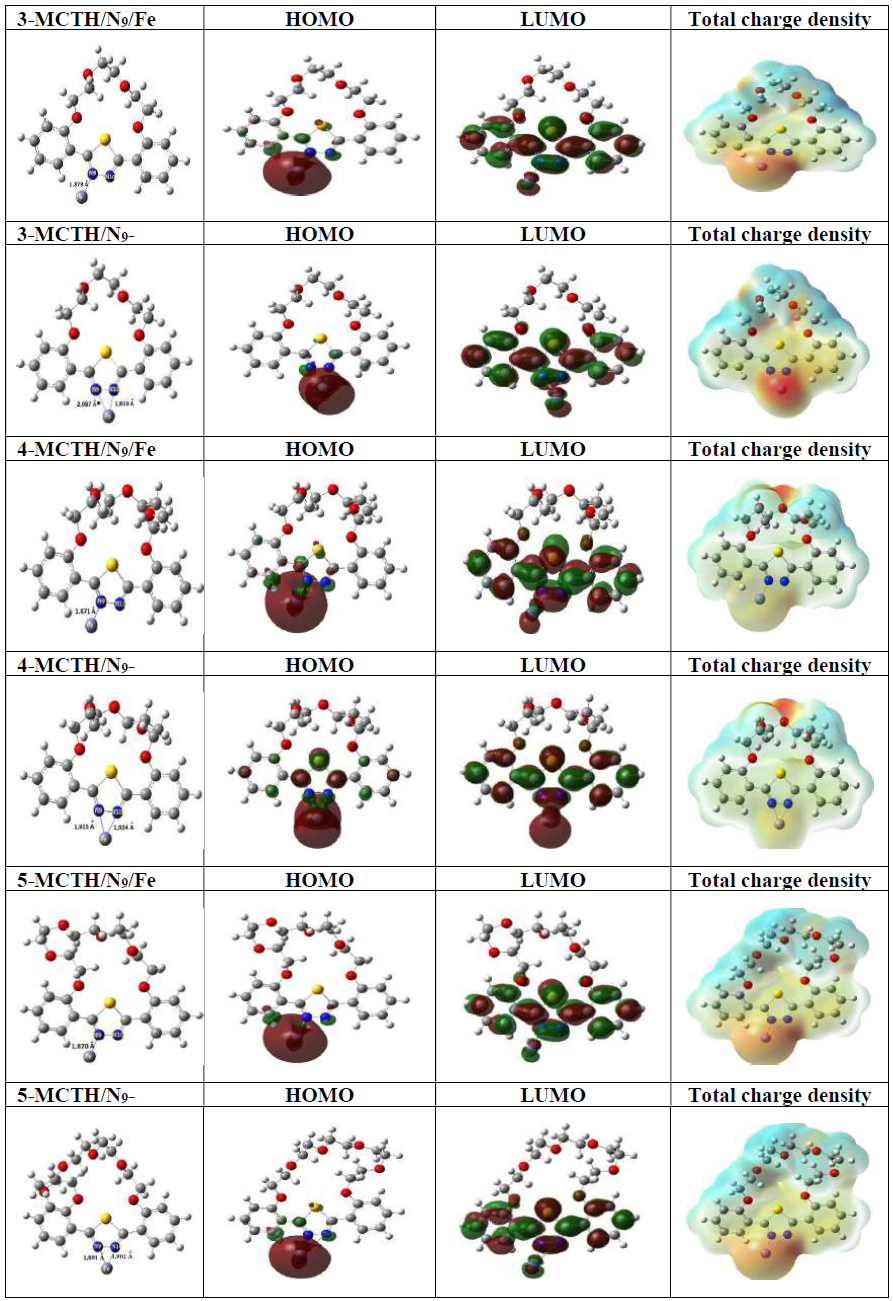
Figure 9 HOMO and LUMO optimized structures and total charge density (T. D.) of the Fe-inhibitor complexes in vacuum .
It is quite clear from Table 10 that the total energies of the neutral and protonated forms in vacuum and aqueous phases increased in the order: 5-MCTH <4-MCTH < 3-MCTH. This ordering indicates that 5-MCTH is the most stable among the three inhibitors. Furthermore, we notice that all the Eads values are negative, which suggests that the neutral and protonated forms can strongly adsorb onto the Fe (111) surface. It should be also noted that 5-MCTH-Fe and its protonated 5-MCTHN9H+N10H+-Fe counterpart have the lowest adsorption energy value in both vacuum (G) and aqueous phases, which confirms the supremacy of their interactions over their 3-MCTH-Fe and 4-MCTH-Fe homologues complexes.
Table 10 Evaluation by Monte Carlo simulation, in vacuum and aqueous phases, of the descriptors of the adsorption configurations onto Fe (111), for 3-MCTH, 4-MCTH and 5-MCTH and their doubly protonated forms at the nitrogen level (all values in kcal / mol).
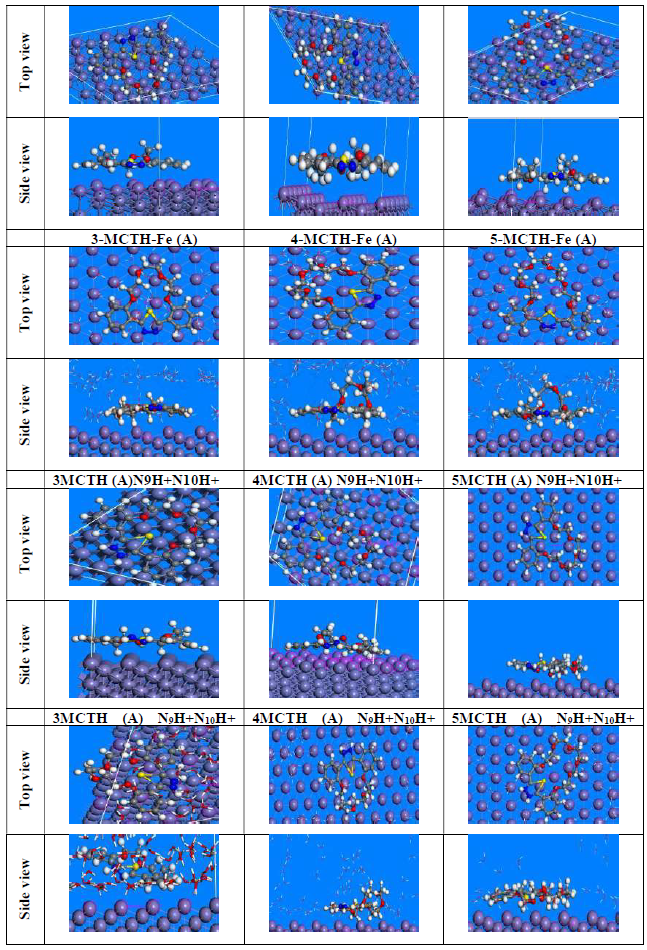
The top and side views of the equilibrium adsorption configurations of 3-MCTH, 4-MCTH and 5-MCTH inhibitors and their protonated forms, onto the Fe (111) surface, in both vacuum (G) and in the aqueous phase (A) (100 H2O), are shown in Fig. 10. Here, we notice easily that the maximum surface recovery is achieved by the parallel adsorption of all inhibitor molecules or protonated forms onto the Fe (111) surface, in both vacuum (G) and the aqueous phases (A). That is why the degree of planarity is a key element in the adsorption efficiency.
From Table 11, collecting the distance between nitrogen atoms and the nearest Fe atom of the Fe (111) surface, it can be emphasized that 5-MCTH, in both neutral and protonated forms, has the lowest distance between nitrogen and the nearest Fe-N surface.
Table 11 Distance between nitrogen atoms and the nearest Fe atom of the Fe (111) surface.
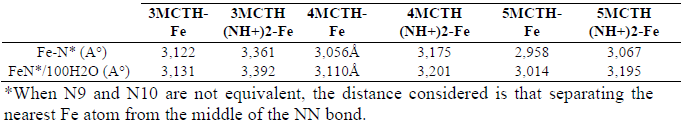
*When N9 and N10 are not equivalent, the distance considered is that separating the nearest Fe atom from the middle of the NN bond.
Conclusion
By combining local and global electronic properties for molecules isolated and in an aqueous solution, we can conclude that 5-MCTH is more reactive in both vacuum and aqueous phase, due to the significant involvement of aryl and thiadiazole rings, in the process of electron donation and acceptance. Moreover, based on the quantum parameters evaluation, the protonated forms are more reactive, in terms of electron acceptance (with a notable superiority of the 5-MCTHN9H +N10H +), than the neutral forms; however, the solvent moderates their reactivity.
In deepening our understanding of the Fe (111) surface complexation mechanism, the results of both DFT and molecular dynamics Monte Carlo approaches showed the importance of the interaction involving a neutral form compared to that of a protonated form, and the supremacy of the interaction of 5-MCTH neutral and protonated variants, compared to their 3-MCTH-Fe and 4-MCTH-Fe complexes homologues.
Authors’ contribution
A. Mahsoune: collected the data; contributed with data or analysis tools; wrote the paper. K. Sadik: contributed with data or analysis tools; performed the analysis; wrote the paper. S. Byadi: collected the data. A. Aboulmouhajir: conceived and designed the analysis; contributed with data or analysis tools; performed the analysis; wrote the paper.

