










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMedicina Interna
versão impressa ISSN 0872-671X
Medicina Interna vol.24 no.2 Lisboa jun. 2017
CASOS CLÍNICOS / CASE REPORTS
Síndrome Hemofagocítica: Um Suspeito a Considerar
Hemophagocytic Syndrome: A Suspect to Have in Mind
Teresa Souto Moura1, Inês Simões1, Marcos Lemos2, Luísa Azevedo1, Rita Gerivaz2, Paula Fonseca1
1Serviço de Medicina 1.4., Hospital de São José, Centro Hospitalar de Lisboa Central, Lisboa, Portugal
2Serviço de Hematologia, Hospital de Santo António dos Capuchos, Centro Hospitalar de Lisboa Central, Lisboa, Portugal
RESUMO
A síndrome hemofagocítica é uma entidade rara e potencial- mente fatal, caracterizada por uma activação descontrolada do sistema imunitário, manifestando-se através de sintomas e sinais clínicos e laboratoriais de inflamação sistémica extrema. Pela inespecificidade dos mesmos, o seu diagnóstico requer um elevado grau de suspeição para implementação de terapêutica adequada e atempada. Os autores apresentam o caso de um jovem de 19 anos, previamente saudável, com febre com três semanas de evolução, interpretada no contexto de infecção respiratória, que subitamente apresentou subida de transaminases e pancitopenia ligeira, exantema cutâneo e hepatoesplenomegália. Os valores elevados de ferritina e receptor solúvel da interleucina-2, bem como a presença de hemofagocitose na medula óssea vieram confirmar o diagnóstico de síndrome hemofagocítica. Após 4 semanas de corticoterapia com dexametasona, assistiu-se a resolução dos sintomas e normalização dos parâmetros laboratoriais.
Palavras-chave:Corticosteroides; Ferritinas/sangue; Linfohistiocitose Hemofagocítica/diagnóstico; Linfohistiocitose Hemofagocítica/tratamento.
ABSTRACT
The hemophagocytic syndrome is a rare and potentially fatal condition, characterized by uncontrolled immune system activation, with symptoms and signs of extreme systemic inflammation. As the manifestations are mostly unspecific, its diagnosis requires a high degree of suspicion, in order to implement adequate and timely therapy. The authors present the case of a 19 year-old boy, previously healthy, with a suspected lung infection due to a period of three weeks fever. He presented a sudden increase of liver enzymes and slight pancytopenia, skin rash and hepatosplenomegaly. The elevated values of ferritin and interleukin-2 soluble receptor, as well as the presence of hemophagocytosis in bone marrow, confirmed the diagnosis of hemophagocytic syndrome. After four weeks of dexamethasone, total resolution of symptoms and laboratorial normalization were achieved.
Keywords:Adrenal Cortex Hormones; Ferritins/blood; Lymphohistiocytosis, Hemophagocytic/diagnosis; Lymphohistiocytosis, Hemophagocytic/therapy
Introdução
A síndrome febril é uma causa comum de recorrência ao médico, um motivo clássico de urgência, consulta ou internamento em Medicina Interna. As suas etiologias são inúmeras, desde situações simples e frequentes, até patologias raras, complexas e cujo diagnóstico constitui um verdadeiro desafio.
Os autores apresentam um caso de febre prolongada em que se diagnosticou uma síndrome hemofagocítica (SHF). Esta é uma doença rara e potencialmente fatal caracterizada por activação descontrolada do sistema imunitário provocando inflamação sistémica extrema e falência multiorgânica.1-3 Inicialmente descrito por Scott e Robb-Smith em 1939 e posteriormente em 1952 por Farquhar e Claireaux que relataram casos fatais de crianças com febre, citopénias progressivas, hepatoesplenomagália e evidência post-mortem de hemofagocitose.4,5 Desde então a mesma constelação de sintomas tem sido descrita tanto em crianças como adultos, com um aumento considerável de casos reportados na literatura.3
Caso Clínico
Jovem do sexo masculino de 19 anos, estudante e trabalhador em restauração em part-time. Sem antecedentes pessoais de relevo ou medicação habitual. Possuía o plano nacional de vacinação actualizado e residia com os pais em ambiente urbano, com boas condições habitacionais e contacto com gato como animal de estimação. Negava tabagismo, admitindo consumo esporádico de canábis inalado e ocasional de álcool ao fim-de-semana. Sem contactos sexuais desprotegidos.
No início de Agosto de 2016, enquanto acampava no Sudoeste Alentejano, iniciou quadro de tosse, expectoração mucosa, odinofagia e mialgias. Recorreu ao médico assistente que o medicou com anti-histamínico e anti-inflamatório, sem melhoria clínica. Por desenvolver febre (temperatura axiar 38ºC), de predomínio nocturno, com sudorese marcada, foi medicado com azitromicina 500 po durante 3 dias, igualmente sem melhoria.
Ao 5º dia de febre, recorreu ao serviço de urgência, onde se apresentava febril (temperatura timpânica 38,4º), taquicardico, normotenso, eupneico em repouso com saturação periférica de oxigénio de 99% em ar ambiente, sem outras alterações significativas ao exame objectivo. Analiticamente apresentava leucocitose 36830/uL, com neutrofilia (33940/ uL), plaquetas 298000/uL, PCR 293 mg/L, função renal, ionograma e enzimologia hepática sem alterações. Perante clínica sugestiva de infecção respiratória com resposta inflamatória evidente, e sem imagens de condensação na teleradiografia de tórax, foi assumida a hipótese de traqueobronquite aguda bacteriana, tendo tido alta medicado com amoxicilina+ácio clavulânico 875/125 mg de 12/12 horas. Por intolerância gastrointestinal ao antibiótico, tendo passado duas semanas após o início dos sintomas, foi internado para terapêutica parentérica - ceftriaxone 2 g/dia – que cumpriu durante 7 dias.
À admissão, encontrava-se febril (38,2ºC), mantendo tosse com expectoração mucosa, sem sintomas evocadores de outro foco infeccioso. Não apresentava alterações cutâneas, nomeadamente exantemáticas ou sugestivas de picada de insecto, adenopatias, organomegálias palpáveis ou sinais meníngeos. A auscultação cardio-pulmonar era inocente, bem como o exame da orofaringe, abdómen e genitais externos. Constatou-se melhoria dos parâmetros de inflamação (PCR 68 mg/L ao 6º dia) mas manteve febre persistente, com vários picos diários > 38,5ºC, de predomínio vespertino, sem cedência aos antipiréticos convencionais (Figura 1). As pesquisas de agente infeccioso (Tabela 1), factor reumatóide, anticorpos antinucleares e anticitrulina revelaram-se negativos. A avaliação imagiológica (tomografia computorizada cervical/tórax/abdominal/pélvico e ecografia abdominal), revelou ligeira hepatomegalia e esplenomegalia ligeira (maior eixo 125 mm), ambos de contornos regulares e ecoestrutura homogénea, sem outras alterações significativas, nomeadamente adenopatias ou colecções abecedadas.
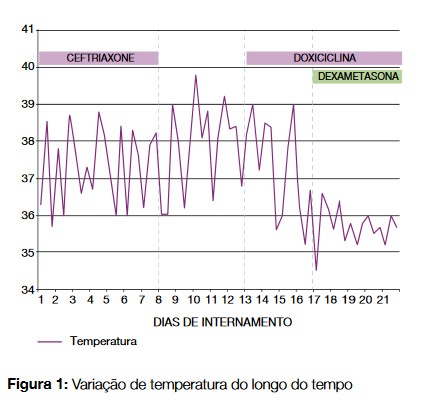
Ao 13º dia de internamento verificou-se aparecimento de exantema macular pruriginoso nos membros e tronco, bem como agravamento analítico com subida de PCR (142 mg/ dL), pancitopenia ligeira (hemoglobina: 11,9 g/dL; leucócitos: 3660/uL; neutrófilos: 2900/uL; plaquetas: 139000/uL) e parâmetros de citocolestase (AST: 201 mg/dL; ALT: 121 mg/ dL, gGT: 442 U/L, fosfatase alcalina:441 U/L) (Figura 2). Foram colhidas novas hemoculturas e iniciada antibioterapia com doxiciclina (200 mg/dia). Perante a febre persistente e as alterações clínicas e analíticas de novo, colocou-se a suspeita de SHF. Para confirmação, foi feito o doseamento de ferritina (máx. 31671 ng/mL [22-275]), fibrinogénio (min. 1,6 g/L [2- 4]), triglicéridos (máx. 235 mg/dL [< 150]) e receptor solúvel da interleucina-2 (sCD25) (9550 pg/mL [< 4800]). Efectuou-se mielograma e biópsia óssea e, após discussão do caso com a Hematologia, decidiu-se iniciar terapêutica com dexametasona. Posteriormente a análise da medula mostrou celularidade aumentada à custa da série granulocitária, observando-se imagens de hemofagocitose no mielograma embora sem tradução na biópsia óssea.
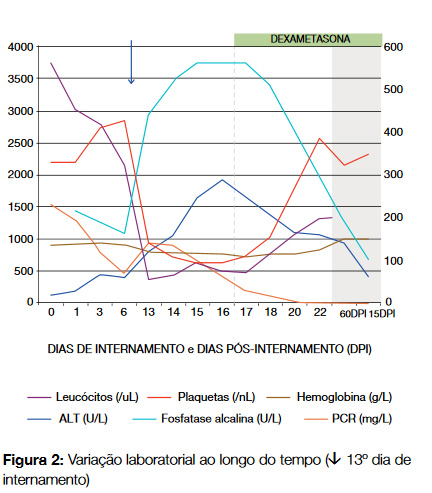
Com o início da dexametasona verificou-se apirexia mantida, resolução do exantema cutâneo e melhoria do estado geral. Laboratorialmente houve normalização das três séries hematológicas e descida dos parâmetros de citocolestase (Figura 2). Ao 22º dia de internamento o doente teve alta, mantendo seguimento em ambulatório, onde se assistiu à normalização da enzimologia hepática e da ferritina às 4 semanas pós internamento. O protocolo de dexametasona durou 8 semanas, com dose inicial 10 mg/m2, e redução para metade a cada 2 semanas. Posteriormente passou a prednisolona 10 mg com desmame gradual durante 4 semanas.
Até à data não se verificou nenhum sinal de recidiva, encontrando-se o doente assintomático.
Discussão
A SHF é classicamente dividida em dois grupos - primária e secundária. A SHF primária ocorre em crianças, tipicamente no primeiro ano de vida, devendo-se a mutações genéticas recessivas que afectam a função do linfócitos T-NK e T-citotóxicos. Nos adultos, a SHF é secundária a uma condição predisponente que está na base da desregulação imune, como neoplasia (especialmente hematológica), doenças auto-imunes ou imunodeficiências e/ou a um trigger como uma infecção bacteriana, viral, fúngica ou parasitária.1,3 Num estudo francês com 162 doentes adultos, a condição mais frequentemente associada à SHF foi a neoplasia (60%), seguida de infecção (25%) e doenças auto-imunes (3%). Dos doentes, 45% encontravam-se imunodeprimidos à apresentação, devido a infecção VIH ou terapêutica imunossupressora.6 Dentro das infecções, o agente, mais frequentemente associado a SHF é o vírus Epstein-Barr. Outros vírus da família herpes, VIH, vírus influenza, Mycobacterium tuberculosis e avium Complex e vários agentes de zoonoses, como Rickettsia, Borrelia e Brucella, são alguns dos microorganismos também implicados.7-9
No presente caso, o factor desencadeante foi provavelmente infeccioso, sendo a neoplasia, a doença auto-imune e a imunossupressão prévias bastante improváveis. Apesar da extensa pesquisa, não foi possível identificar o microorganismo responsável, tendo-se optado inicialmente por uma cefalosporina e mais tarde, considerando a epidemiologia, pela doxiciclina para garantir cobertura de Rickettsias, Coxiella burnetti, Borrelia e Leptospira. O que despertou a hipótese de SHF foi o aparecimento súbito de citopénias e citocolestase em doente com febre elevada há três semanas, exantema cutâneo e hepatoesplenomegália, ainda que ligeira. Esta foi posteriormente corroborada pelos valores invulgarmente elevados de ferritina e de sCD25, bem como pela presença de imagens de hemofagocitose no mielograma.
Pela inexistência de sintomas ou sinais específicos o diagnóstico da SHF pode ser desafiante. Em 2007 a Histiocyte Society publicou os critérios utilizados no estudo HLH-2004 (Tabela 2) que incluem a identificação de mutações genéticas associadas à SHF ou cinco dos oito seguintes critérios: febre, esplenomegalia, citopénias, hipertrigliceridémia ou hipofibrinogenémia, evidência de hemofagocitose, hiperferritinémia,actividade diminuída das células NK e aumento do sCD25. Outros achados consistentes com o diagnóstico são sintomas meníngeos, adenomegalias, icterícia, edema, exantema cutâneo, aumento da enzimologia hepática, hipoproteinémia, hiponatrémia, aumento de LDL e diminuição de HDL.10
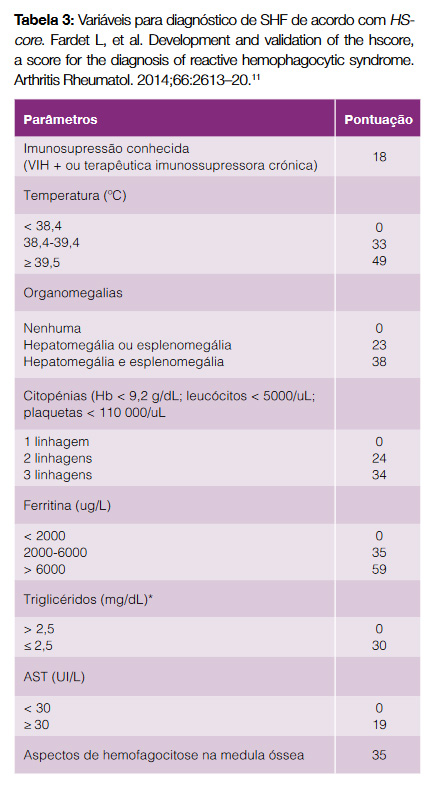
Ainda que amplamente utilizados, estes critérios foram concebidos para crianças, não havendo até à data uma adaptação consensual para adultos; exigem acesso a testes genéticos, actividade das células NK e doseamento de sCD25, que não estão universalmente disponíveis; e nem todos os critérios estão presentes no momento de apresentação da doença, tornando fundamental um elevado nível de suspeição.1,3 Em alternativa, Fardet et al propuseram novos critérios para a SHF secundária, com validação em adultos, denominados HScore (Tabela 3).11 A cada parâmetro é atribuída uma pontuação, cuja soma corresponde a uma probabilidade de SHF – no presente caso foi de 99%.
O sCD25 não está disponível em todos os hospitais, motivo pelo qual foi retirado do HScore, mas além de útil para o diagnóstico, correlaciona-se com a actividade da doença mais do que outros biomarcadores.2 A ferritina é também um marcador de inflamação relevante, como exposto num estudo pediátrico, em que valores de ferritina > 10000 ug/L demonstraram sensibilidade de 90% e especificidade de 96% para SHF.12 Contrariamente, na população adulta, a elevação da ferritina não é específica de SHF, não existindo até à data um cut-off consensual.13 Ainda que a presença de hemofagocitose seja a marca da SHF, esta não é específica, estando presente em situações frequentes como cirurgias, sépsis ou transfusões. Por outro lado, vários estudos mostraram que a prevalência de hemofagocitose na medula óssea de doentes com SHF se situa entre 25-100%, não sendo indispensável para o diagnóstico a sua demonstração histológica.3,6,14,15
O tratamento pressupõe, por um lado, a interrupção do factor desencadeante da SHF (infeccioso, neoplásico ou auto-imune) e por outro, o controlo do sistema imunitário desregulado. A corticoterapia é a base da terapêutica, utilizando-se adicionalmente protocolos baseados em etoposido (recomendações do HLH-2004) ou CHOP (ciclofosfamida, doxirrubicina, vincristina e prednisona) se houver necessidade de fármacos citotóxicos.1,10 No entanto, não há consenso quanto à terapêutica inicial ideal bem como a sua duração. O presente caso, por ter sido diagnosticado e tratado precocemente, foi abordado exclusivamente com corticoterapia, com excelentes resultados. A antibioterapia empírica instituída terá sido também relevante para o tratamento, ao controlar a infecção considerada como desencadeante provável da SHF.
O caso apresentado pretende ilustrar uma entidade rara, porém real e provavelmente subdiagnosticada. Pela inespecificidade dos sintomas e sinais associados a um prognóstico reservado, se não tratado, a SHF exige de qualquer internista, um índice de suspeição elevado para um diagnóstico e tratamento atempados.
Referencias
1.Hayden A, Park S, Giustini D, Lee AY, Chen LY. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev. 2016;30:411-20. [ Links ]
2.Jordan MB, Allen CE, Weitzman S, Filipovich AH, Mcclain KL. How I treat How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041– 52. [ Links ]
3.Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2016;125:2908–15. [ Links ]
4.Scott R,Robb-Smith A. Histiocytic medullary reticulosis.Lancet.1939;234:194–8. [ Links ]
5.Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27:519–25. [ Links ]
6.Rivière S, Galicier L, Coppo P, Marzac C, Aumont C, Lambotte O, et al. Reactive hemophagocytic syndrome in adults: A retrospective analysis of 162 patients. Am J Med. 2014;127:1118–25. [ Links ]
7.Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814–22. [ Links ]
8.Maakaroun NR, Moanna A, Jacob JT, Albrecht H. Viral infections associated with haemophagocytic syndrome. Rev Med Virol. 2010;20:93–105. [ Links ]
9.Cascio A, Pernice LM, Barberi G, Delfino D, Biondo C, Beninati C, et al. Secondary hemophagocytic lymphohistiocytosis in zoonoses. A systematic review. Eur Rev Med Pharmacol Sci. 2012;16:1324–37. [ Links ]
10.Henter J, Horne A, Aricó M, Egeler R, Filipovich A, Imashuku S, et al. HLH-2004 : Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. [ Links ]
11.Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the hscore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613– 20. [ Links ]
12.Allen CE, Yu X, Kozinetz CA, McClain KL. Highly Elevated Ferritin Levels and the Diagnosis of Hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer. 2008;50:1227–35. [ Links ]
13.Schram AM, Campigotto F, Mullally A, Fogerty A, Massarotti E, Neuberg D, et al. Marked hyperferritinemia does not predict for HLH in the adult population. Blood. 2015;125:1548–52. [ Links ]
14.Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol. 2014;141:62–71. [ Links ]
15.Gupta A, Weitzman S. The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50:192–4. [ Links ]
Correspondência: Teresa Souto Moura teresasoutomoura@gmail.com
Serviço de Medicina 1.4., Hospital de São José,
Centro Hospitalar de Lisboa Central, Lisboa,
Portugal Rua José António Serrano, 1150-199 Lisboa
Protecção de Seres Humanos e Animais: Os autores declaram que não foram realizadas experiências em seres humanos ou animais.
Direito à Privacidade e Consentimento Informado: Os autores declaram que nenhum dado que permita a identificação do doente aparece neste artigo.
Conflitos de Interesse: Os autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
Fontes de Financiamento: Não existiram fontes externas de financiamento para a realização deste artigo.
Recebido: 31/10/2016
Aceite: 27/12/2016


{kind=link}