










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMedicina Interna
versão impressa ISSN 0872-671X
Medicina Interna vol.25 no.1 Lisboa mar. 2018
ARTIGOS ORIGINAIS / ORIGINAL ARTICLES
T-Cell Large Granular Lymphocyte Leukemia: The Experience of a Single Center
Leucemia de Grandes Linfócitos Granulares de Células T: A Experiência de uma Instituição
Isabel M. Eira1, Sandra I. Correia1, Cristina Ângela1, Herlander Marques2,3
1Internal Medicine Department, Hospital de Braga, Braga, Portugal
2Oncology Department, Hospital de Braga, Braga, Portugal
3Life and Health Sciences Research Institute (ICVS), School of Health Sciences, University of Minho, Braga, Portugal
ABSTRACT
Background: T-cell large granular lymphocyte (LGL) leukemia is a rare lymphoproliferative disease. It frequently involves the expansion of CD8+ cells, which may lead to cytopenias and often correlates with autoimmune disorders. Another form is the CD4+ LGL leukemia, which behaves more indolently but may associate with other neoplasia.
Material and Methods: Retrospective and descriptive analysis of the 14 patients diagnosed with T-cell LGL leukemia in our center between 2002 and 2016, regarding gender/age; clinical presentation; immunophenotype; frequency of cytopenias; coexistent malignancies; autoimmune disorders and temporal relationship between both diagnosis; immunosuppressant treatment and its outcome.
Results: Three patients had CD4+ LGL leukemia and eleven had CD8+ LGL leukemia. In the latter, neutropenia was the prevailing cytopenia (63.6%), followed by anemia (45.5%) and thrombocytopenia (36.4%). The most common symptoms were fatigue and recurrent bacterial infections; 35.7% presented with asymptomatic cytopenias. From patients with CD4+ LGL leukemia, one had colorectal cancer. Prevalence of autoimmune diseases was 35.7%; its diagnosis preceded the identification of LGL leukemia in all patients. Five patients required treatment; methotrexate was the most selected firstline immunosuppressant. Complete hematological response was achieved in two cases. Mortality rate was 14.3% at a median follow-up time of 2 years.
Discussion and Conclusion: Clinical presentation and frequency of cytopenias were close to described in previous studies. The recurrent association with autoimmunity suggests the existence of common etiopathogenic features; chronic autoantigen stimulation might play a role in the onset of the clonal disease. Further studies are needed for determining the gold-standard approach of LGL leukemia.
Keywords: Autoimmune Diseases; Leukemia, Large Granular Lymphocytic; Lymphoma, T-Cell.
RESUMO
Introdução: A leucemia de grandes linfócitos granulares (LGL) de células T é uma doença linfoproliferativa rara. Esta envolve frequentemente a expansão de células CD8+, podendo levar a citopenias e correlacionar-se com distúrbios autoimunes. Outra forma, a leucemia LGL-CD4+, apresenta um comportamento mais indolente, mas pode associar-se a outras neoplasias.
Material e Métodos: Estudo retrospetivo e descritivo dos 14 doentes diagnosticados com leucemia LGL de células T no nosso centro entre 2002 e 2016, avaliando: género/idade; apresentação clínica; imunofenotipagem; frequência de citopenias; neoplasias coexistentes; doenças autoimunes e relação temporal entre diagnósticos; tratamento imunossupressor e seu resultado.
Resultados: Três doentes tinham leucemia LGL-CD4+ e onze tinham leucemia LGL-CD8+. Nestes últimos, a neutropenia foi a citopenia mais prevalente (63,6%), seguida da anemia (45,5%) e trombocitopenia (36,4%). Os sintomas mais frequentes foram fadiga e infeções bacterianas recorrentes; 35,7% apresentavam citopenias assintomáticas. Dos doentes com leucemia LGL-CD4+, um apresentava neoplasia do cólon. A prevalência de doenças autoimunes foi 35,7%; este diagnóstico precedeu sempre o da leucemia. Cinco doentes iniciaram terapêutica; o metotrexato foi o imunossupressor de primeira linha mais utilizado. Verificou-se resposta hematológica completa em dois casos. A taxa de mortalidade foi de 14,3%, num seguimento mediano de 2 anos.
Discussão e Conclusão: A apresentação clínica e a frequência de citopenias foram semelhantes ao descrito em estudos anteriores. A recorrente associação com autoimunidade sugere a existência de mecanismos etiopatogénicos comuns; a estimulação crónica por autoantigénios poderá desempenhar um papel no desencadear da doença clonal. São necessários mais estudos para determinar a abordagem gold-standard desta patologia.
Palavras-chave: Doenças Autoimunes; Leucemia Linfocítica Granular de Células Grandes; Linfoma de Células T.
Introduction
Large granular lymphocytes (LGL) are a morphologically distinct subpopulation with abundant cytoplasm and azurophilic granules, that normally comprises 10% to 15% of peripheral blood mononuclear cells.1,2 The normal LGL count in peripheral blood is 0,25 × 109/L.3 These lymphocytes are classified into two distinct lineages as either CD3+ T cells or CD3- natural killer (NK) cells, both types playing important roles in the immune system.
LGL leukemia embodies a spectrum of rare clonal lymphoproliferative diseases involving inappropriate expansion of LGL. T-cell LGL leukemia represents the most frequent LGL disorder, accounting for 85% of all cases.1 In T-cell LGL leukemia, the abnormally expanded clone usually bears the phenotype of mature effector CD8+ cytotoxic T cells.4,5 CD8+ LGL leukemia is associated to a mild to moderate lymphocytosis, neutropenia, anemia and/or thrombocytopenia. This disorder is more frequent in the elder and both genders are equally affected.1
Recently there have been reports about CD4+ LGL leukemia, with differences from the classical CD8+ LGL leukemia. It shows an indolent course in the absence of cytopenia or symptoms attributed to the lymphoproliferative disorder itself; however, CD4+ LGL leukemia frequently have a higher incidence of an associated second neoplasia, which usually determines the clinical course of the disease.4
LGL cells have a mature, post-thymic phenotype.6,7 As the lymphocytes rearrange their T-cell receptor (TCR) genes during normal development, all cells arising from a malignant, transformed T cell will have the same sequence of TCR.1 About 90% of patients with T-cell LGL leukemia express the heterodimer TCR-aß; the minority of patients expressing TCR-?d seem to be associated with a more favorable prognosis.6
Diagnosis of T-cell LGL leukemia is based on finding a persistent (> 6 months) expansion of LGL in peripheral blood (> 0.5 × 109/L), with typical morphology on the peripheral smear; an expanded LGL population with a characteristic immunophenotype by flow cytometry; and evidence of T-cell clonality by the detection of a clonal rearrangement of the TCR gene by polymerase chain reaction (PCR) or by flow cytometry.3,5,6,8
Clinical features of LGL leukemia include recurrent bacterial infections, fatigue and rarely B symptoms (fever, night sweats and weight loss).9 More than a third of patients come to medical attention with asymptomatic cytopenias. Neutropenia is found in 60 to 85% of cases (50% of them with severe neutropenia); anemia and thrombocytopenia are also frequent (48% and 20% of cases, respectively).3,10,11 Splenomegaly is observed in 25% to 50% of cases, while hepatomegaly and lymphadenopathies are rarely found.3 Although the clinical behavior is typically indolent, about 60% to 70% of patients will become symptomatic at some point in the disease course and will require specific therapy.8
One of the unique features of T-cell LGL leukemia is its common association with other clinical conditions such as autoimmune diseases, further hematological disorders and other malignancies.1 Autoimmune disorders are thought to be present in up to 74% of patients with LGL leukemia, including arthropathies, vessel and skin disorders, neuropathies, muscular and glandular diseases, and other connective tissue disorders.2,8 Association with rheumatoid arthritis (RA) is the most recognized; it is reported in about one third of LGL leukemia patients. Signs and symptoms of autoimmune conditions can present before the features of LGL leukemia become evident.8 This correlation is not yet completely understood and has led to the hypothesis that T-cell LGL leukemia arises on a background of sustained immune stimulation.12
The frequency of LGL leukemia is not accurately determined and ranges from 2% - 5% of chronic lymphoproliferative disorders in North America and up to 5% - 6% in Asia.3,6 The first population-based study was recently published, and confirmed that LGL leukemia is an extremely rare disease, with an annual incidence of 0.2 cases per million individuals in the United States.13 In Europe, there are no available data about the frequency of LGL leukemia; the largest published series consists of 229 patients and was conducted in France.14
In the present study, we aim to characterize the patients diagnosed with T-cell LGL leukemia in our center, with focus to the clinical course, relationship with autoimmune disorders and treatment options.
Material and Methods
A retrospective and descriptive analysis was conducted, by revision of the clinical records of the patients diagnosed with T-cell LGL leukemia in our center between the years of 2002 and 2016.
Inclusion criteria were based on clinical and laboratorial data. It included patients aged 18 years or older who met T-cell LGL leukemia diagnostic criteria: an expanded LGL population with a characteristic immunophenotype by flow cytometry and evidence of a clonal rearrangement of the TCR gene in peripheral blood or bone marrow aspirate by PCR (with probes to rearrangement of TCR-ß chain or ? chain). The standard set of florescent-labeled antibodies included CD3, CD4, CD5, CD8, CD20, CD23, CD38, CD79a, TCR-aß and TCR-?d.
Patients with NK-LGL chronic lymphoprolipherative disease were excluded.
An analysis was made to characterize the 14 patients who met the inclusion criteria regarding: gender and age; clinical presentation; immunophenotype; frequency of cytopenias; coexistent malignancies; coexistent autoimmune disorders and temporal relationship between both diagnosis; immunosuppressant treatment and its outcome.
The diagnosis of autoimmune diseases was based on clinical features and detection of autoantibodies. The diagnosis of associated malignancy was obtained by histological evaluation of neoplastic tissue. Standard imaging techniques were used to staging procedures.
Results
There were 14 cases of T-cell LGL leukemia registered in our center; the diagnosis was made between the years of 2002 and 2016. The median follow-up time was 2 years (ranging from 1 to 14 years). Demographic features of the patients are exposed in Table 1. Most of the patients were female (n = 12, 85.7%). The mean age at diagnosis was 66.9 ± 10.8 years. Two patients (14.3%) were diagnosed under the age of 50 years.
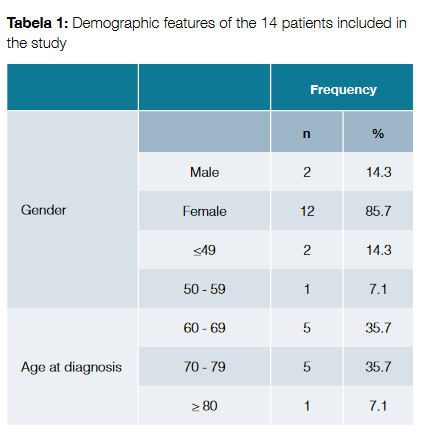
Clinical features at presentation are exposed in Table 2. The most common symptom was fatigue, followed by recurrent bacterial infections. On the other hand, 35.7% of patients presented with asymptomatic cytopenias.
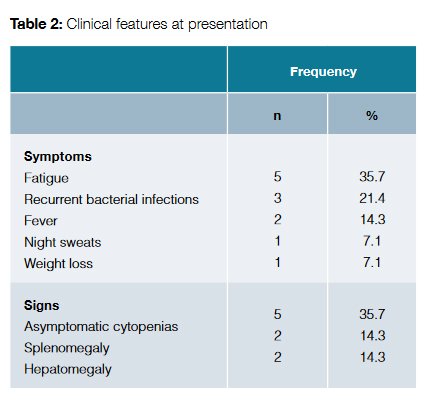
CD8+ T-cell LGL leukemia was present in 11 of the cases (78.6%). From these, nine patients (81.8%) exhibited an expansion of the TCR-aß gene; the other two patients (18.2%) had a rearrangement of the TCR-?d gene.
Three patients (21.4%) had CD4+ T-cell LGL leukemia. Two of them (66.7%) were asymptomatic and had no laboratorial anomalies. The third one had concomitant diagnosis of metastatic colorectal cancer and was the only patient with CD4+ T-cell LGL leukemia who had neutropenia and anemia.
Regarding hematological involvement in patients with CD8+ T-cell LGL leukemia, neutropenia was the most frequent cytopenia, being present in seven patients (63.6% of the cases). From these, four patients (57.1%) had severe neutropenia (absolute neutrophil count <500/µL). Anemia was detected in five patients (45.5%) and was severe in 80.0% of the cases (hemoglobin < 8 g/dL). One of the patients had autoimmune hemolytic anemia. Thrombocytopenia was present in four patients (36.4%) and was mild to moderate (platelet count > 50 000/uL).
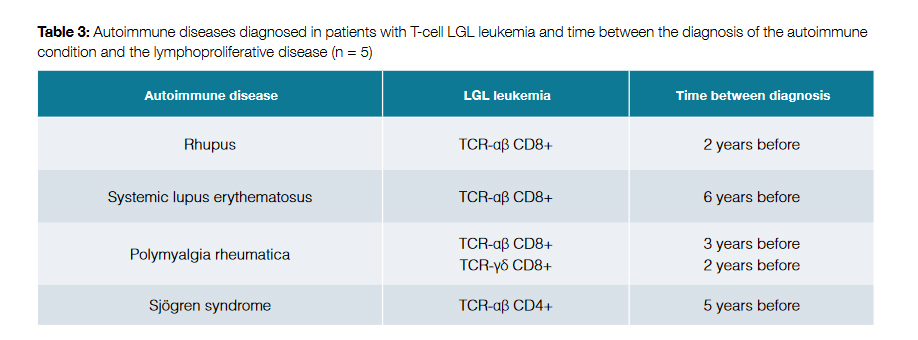
The prevalence of autoimmune diseases in patients with T-cell LGL leukemia was 35.7%. The diagnosis of the autoimmune condition preceded the identification of the lymphoproliferative disorder in all cases, in an average time of 3.6 ± 1.8 years. Table 3 shows the identified pathologies and its relationship with the LGL leukemia.
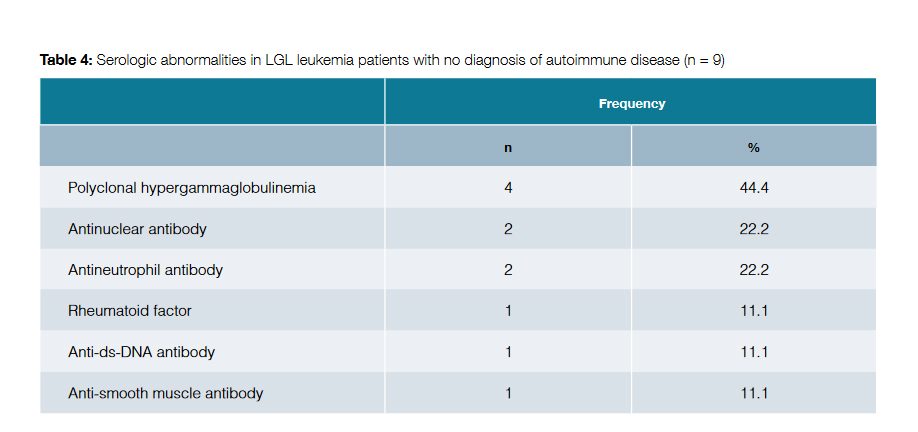
The remaining patients with T-cell LGL leukemia did not meet diagnostic criteria for any autoimmune disorder. However, elevated titers of various autoantibodies were documented in some of these patients, as shown in Table 4.
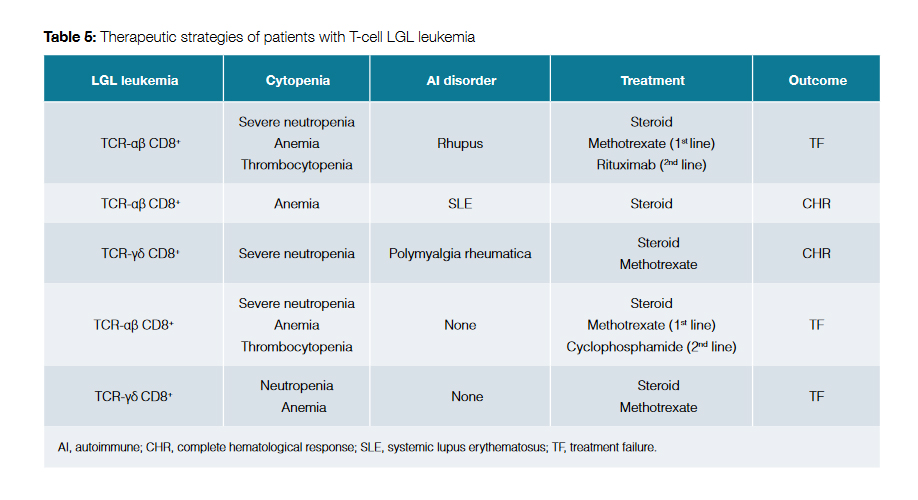
Pharmacological therapy was started in five of the cases (35.7%), all of them with CD8+ T-cell LGL leukemia. Table 5 shows the features of the treated patients, the therapeutic strategies and respective outcomes.
Methotrexate was selected as first-line therapy in four cases: three of them because of severe neutropenia and the other one because of transfusion dependent anemia. Adjunctive steroid therapy was used in all these patients. One patient had complete hematological response with methotrexate, resulting in a 25.0% complete response rate. The other three patients experienced treatment failure.
One of the patients who failed to respond to methotrexate had concomitant diagnosis of rhupus (an overlapping syndrome with features of RA and systemic lupus erythematosus (SLE)). This patient posteriorly underwent therapy with rituximab but did not respond as well; the patient ultimately died because of an infectious intercurrence related with severe neutropenia.
Other patient underwent second line therapy with oral cyclophosphamide but there was no response to this therapy either, and the patient is now under symptomatic treatment.
The third patient who failed to respond to methotrexate did not undergo second line therapy and is also under symptomatic treatment.
A patient with concomitant diagnosis of SLE and transfusion dependent anemia experienced complete hematologic response only with steroid therapy.
At a median follow-up time of 2 years (ranging from 1 to 14 years), 2 patients have died, resulting in a mortality rate of 14.3%. One of the patients had CD8+ T-cell LGL leukemia with severe neutropenia, transfusion dependent anemia and an associated autoimmune disorder (rhupus), and died 3 years after diagnosis because of an infectious intercurrence. The other patient had CD4+ T-cell LGL leukemia and died 2 years after diagnosis; the fatal outcome was dictated by the concomitant diagnosis of metastatic colorectal adenocarcinoma.
Discussion
LGL leukemia is thought to be a disease of the elderly, presenting typically in the sixth decade of life; in fact, the mean age at the time of diagnosis in our series was 66.9 years. However, in contrast to the equal incidence between genders described in previous studies, female patients were significantly more affected in our series (85.7% of patients).
Regarding clinical features at presentation, fatigue and recurrent bacterial infections were the most reported symptoms, which can be mainly explained by the presence of anemia and severe neutropenia, respectively. The fraction of patients who came to medical attention because of asymptomatic cytopenias (35.7%) was very close to data from previous reviews, which reported that this occurs in about one third of the patients.1
Concordant with previous studies, the most common immunophenotype was the CD8+ LGL leukemia, present in 78.6% of patients.
CD4+CD8-/+dim LGL leukemia is described as having clear clinical differences from the classical CD8+ LGL leukemia, such as the absence of cytopenias and its association with other neoplasia in about one third of the cases.4,15 In fact, previous studies suggest that the presence of clinical symptoms and signs or laboratory anomalies in patients with CD4+ LGL leukemia should be considered as an alert for a possible associated neoplasm.4 In our study, there were three cases of CD4+ LGL leukemia. Two of the patients were asymptomatic and had no laboratory anomalies. The other patient had concomitant diagnosis of metastatic colorectal cancer, which was responsible for the unfavorable outcome.
Although most cases of LGL leukemia behave in an indolent manner, many patients become symptomatic during the course of the disease.11 Neutropenia is the most common hematologic anomaly in LGL leukemia and is related with several mechanisms, including the direct inhibition of myeloid precursors by infiltrating leukemic cells, dysregulation of the maturation of myeloid cells, antibody- or immune complex-mediated neutrophil destruction, hypersplenism, and Fas/ Fas-ligand induced premature apoptosis of neutrophils.10,11 In our study, neutropenia was present in 63.6% of CD8+ LGL leukemia cases, concordant with previous data (60% to 85%). The fraction of cases with severe neutropenia was 57.1%, a frequency similar with the described in previous studies.
Anemia is reported to occur in 48% of patients with CD8+ LGL leukemia, because of cytotoxic activity of the clonal large lymphocytes.8,10 The frequency of anemia in our study was very close to this number (45.5%). When present, anemia was severe in 80.0% of the cases. In our series, one of the patients had Coombs-positive autoimmune hemolytic anemia, which only occurs in rare cases.10
Thrombocytopenia may be related with the inhibition of megakaryopoiesis in bone marrow by LGL leukemic cells, antibody-mediated peripheral destruction and splenic sequestration.8,10 In our study, thrombocytopenia was present in 36.4% of patients with CD8+ LGL leukemia, a percentage slightly above the reported 20%. There were no cases of severe thrombocytopenia.
T-cell LGL leukemia is known to be associated with a wide spectrum of systemic autoimmune diseases, with most of them involving connective tissue.2,8 Dysregulation of the normal function of B-cells due to the presence of malignant LGLs and production of proapoptotic and proinflammatory cytokines are thought to be responsible for increased autoimmunity in these patients.11 In our study, the prevalence of clinical autoimmune syndromes in patients with T-cell LGL leukemia was 35.7%. In patients with CD8+ LGL leukemia, there were two cases of polymyalgia rheumatica, one case of rhupus (an overlapping syndrome with features of RA and SLE) and one case of SLE. Although previous studies did not find an association between CD4+ LGL leukemia and autoimmune diseases4, there was one case of Sjögren syndrome in a patient with CD4+ LGL leukemia in this series.
Patients with T-cell LGL leukemia may present with signs and symptoms of autoimmune diseases before the manifestations of leukemia become evident.8 In fact, in our study, the diagnosis of the autoimmune condition preceded the identification of the lymphoproliferative disorder in all cases, in an average time of 3.6 ± 1.8 years. This might support the alternative theory that the autoimmune conditions may be primary and even play a role in the onset of the clonal disease.8
Elevated titers of various autoantibodies have been identified in patients with T-cell LGL leukemia.8 In our study, in the patients with no diagnosis of autoimmune disorder, polyclonal hypergammaglobulinemia was present in 44.4% of cases; positivity to antinuclear antibody, antineutrophil antibody, rheumatoid factor, anti-ds-DNA antibody and anti-smooth muscle antibody was also identified in smaller percentages. The presence of these autoantibodies may identify subjects in risk for autoimmunity, individuals in the process of developing an autoimmune condition, or even patients with an undiagnosed autoimmune disorder.8
Systemic immunosuppressive therapy is the foundation of treatment of both LGL leukemia and autoimmune disorders. When these entities coexist, most authors recommend starting treatment with methotrexate, which controls both rheumatologic symptoms and cytopenias.3 Nonetheless, cyclophosphamide and cyclosporine are also acceptable options for treatment of LGL leukemia patients with autoimmune diseases.8
T-cell LGL leukemia pursue a chronic clinical course and, in asymptomatic patients, a watch and wait approach should be adopted.3 Indications for starting therapy are: a) severe neutropenia; b) moderate neutropenia symptomatic from recurrent infections; c) symptomatic or transfusion dependent anemia; and d) associated autoimmune conditions requiring treatment.3,6
Primary response is evaluated 4 months after starting therapy and can be classified into: complete hematologic response (normalization of blood counts and circulating LGL in the normal range); partial hematologic response (improvement in blood counts that do not meet criteria for complete remission); or treatment failure (any response not meeting the previous criteria).3
First-line agents for the treatment of LGL leukemia are methotrexate or cyclophosphamide. Prednisone may be used as an adjunct therapy if absolute neutrophil count is under 200/µL or in the setting of an autoimmune disease. In case of treatment failure, switching to methotrexate/cyclophosphamide/cyclosporine (according to first choice therapy) is indicated. Cyclosporine is a possible second-line therapy in case of anemia. Both methotrexate and cyclosporine can be maintained indefinitely if tolerated; cyclophosphamide therapy is limited to 6 to 12 months because of the risk of bladder toxicity and mutagenesis. Refractory cases are defined as failure of the 3 major immunosuppressive drugs. For these patients, options include purine analogs, alemtuzumab and splenectomy, with variable results.3
Methotrexate is the initial choice for LGL leukemia patients with neutropenia and/or an associated autoimmune disease; it is also an option for first-line therapy of LGL leukemia patients with anemia.3 In previous series, overall response rate achieved with methotrexate was 55%, while complete response rate was 21%.12 In our study, four patients were treated with methotrexate and complete hematological response was achieved in only one case.
Cyclophosphamide is an alternative first-line therapy for LGL leukemia patients with anemia.3 Previous data reported a 66% overall response rate and a 47% complete response rate.12 In our study, cyclophosphamide was used in only one patient and as second line therapy; it resulted in treatment failure as well.
Adjunctive steroid therapy may provide a more rapid hematological improvement while first-line immunosuppressive agents are being initiated.3 Previous data evidenced limited efficacy of monotherapy with steroids, with 12% overall response rate and 3% complete response rate.12 In our study, a patient with concomitant diagnosis of SLE and transfusion dependent anemia had a complete hematological response only with steroid therapy.
Recent reviews of clinical cases reported a long-term remission of LGL leukemia in patients with concomitant diagnosis of RA treated with rituximab.16,17 The paradoxical efficacy of this specific anti-B-cell therapy on a T-cell monoclonal disease raised the theory that, in some cases, LGL leukemia may be a reactive manifestation of chronic autoantigen stimulation rather than a true malignancy.16 In our study, one patient with LGL leukemia and rhupus underwent second-line therapy with rituximab after treatment failure with methotrexate. However, there was no hematological response to this treatment as well.
T-cell LGL leukemia is a chronic disease with unclear impact on survival; previous studies showed a median survival of 9 to 10 years.10,13 In our study, the mortality rate at a median follow-up time of 2 years (ranging from 1 to 14 years) was 14.3%. Further studies are required to understand its prognosis.
Conclusion
T-cell LGL leukemia is a rare clonal disease that most commonly affects the elderly population. A correct definition of the malignant cells phenotype is important in predicting significant clinical features and guiding the appropriate approach. In the latest years, significant advances in understanding the dysregulation of multiple survival pathways involved in the pathophysiology of this disease have been made, putting LGL leukemia in the intersection of malignancy and autoimmunity. As none of the current therapeutic modalities can be curative, further studies are needed for determining the gold-standard approach of T-cell LGL leukemia. The understanding of these mechanisms may be important for the development of new treatment options.
Referencias
1. Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist. 2004;9:247-58. [ Links ]
2. Zhang R, Shah M, Loughran TP Jr. The root of many evils: indolent large granular lymphocyte leukemia and associated disorders. Hematol Oncol. 2010;28:105-17. [ Links ]
3. Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood. 2011;117:2764-74. [ Links ]
4. Lima M, Almeida J, Teixeira MA, Alquero MC, Santos AH, Balanzategui A, et al. TCRαβ+/CD4+ large granular lymphocytosis: a new clonal T-cell lymphoproliferative disorder. Am J Pathol. 2003;163:763-71.
5. Shah A, Diehl LF, St Clair EW. T cell large granular lymphocyte leukemia associated with rheumatoid arthritis and neutropenia. Clin Immunol. 2009;132:145–52.
6. Steinway SN, LeBlanc F, Loughran TP Jr. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev. 2014;28:87–94.
7. LeBlanc F, Zhang D, Liu X, Loughran TP Jr. Large granular lymphocyte leukemia: from dysregulated pathways to therapeutic targets. Future Oncol. 2012;8:787-801. [ Links ]
8. Bockorny B, Dasanu CA. Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk. 2012;12:400-5. [ Links ]
9. Morice WG, Kurtin PJ, Tefferi A, Hanson CA. Distinct bone marrow findings in T-cell granular lymphocytic leukemia revealed by paraffin section immunoperoxidase stains for CD8, TIA-1, and granzyme B. Blood. 2002;99:268-74. [ Links ]
10. Sokol L, Loughran TP Jr. Large granular lymphocyte leukemia. Curr Hematol Malig Rep. 2007;2:278-82. [ Links ]
11. Alekshun TJ, Sokol L. Diseases of large granular lymphocytes. Cancer Control. 2007;14:141-50. [ Links ]
12. Dearden CE, Johnson R, Pettengell R, Devereux S, Cwynarski K, Whittaker S, et al. British Committee for Standards in Haematology. Guidelines for the management of mature T-cell and NK-cell neoplasms (excluding cutaneous T-cell lymphoma). Br J Haematol. 2011;153:451–85.
13. Shah MV, Hook CC, Call TG, Go RS. A population-based study of large granular lymphocyte leukemia. Blood Cancer J. 2016;6:e455. [ Links ]
14. Bareau B, Rey J, Hamidou M, Donadieu J, Morcet J, Reman O, et al. Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica. 2010;95:1534–41.
15. Garrido P, Ruiz-Cabello F, Bárcena P, Sandberg Y, Cantón J, Lima M, et al. Monoclonal TCR-Vβ13.1+/CD4+/NKa+/CD8-/+dim T-LGL lymphocytosis: evidence for an antigen-driven chronic T-cell stimulation origin. Blood. 2007;109:4890-8.
16. Cornec D, Devauchelle-Pensec V, Jousse-Joulin S, Marhadour T, Ugo V, Berthou C, et al. Long-term remission of T-cell large granular lymphocy Table 5: Therapeutic strategies of patients with T-cell LGL leukemia. te leukemia associated with rheumatoid arthritis after rituximab therapy. Blood. 2013;122:1583-6 [ Links ]
Correspondência: Isabel M. Eira isi.morgado@gmail.com
Internal Medicine Department, Hospital de Braga, Braga, Portugal
Sete Fontes - São Victor, 4710-243 Braga
Presentations / Apresentações: A part of this work was presented at the XXII National Congress of Internal Medicine.
Conflicts of interest: The authors have no conflicts of interest to declare.
Financing Support: This work has not received any contribution, grant or scholarship.
Confidentiality of data: The authors declare that they have followed the protocols of their work center on the publication of data from patients.
Protection of human and animal subjects: The authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Conflitos de Interesse: Os autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
Fontes de Financiamento: Não existiram fontes externas de financiamento para a realização deste artigo.
Proteção de Pessoas e Animais: Os autores declaram que os procedimentos seguidos estavam de acordo com os regulamentos estabelecidos pelos responsáveis da Comissão de Investigação Clínica e Ética e de acordo com a Declaração de Helsínquia da Associação Médica Mundial.
Confidencialidade dos Dados: Os autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de doentes.
Recebido: 19/07/2017
Aceite: 15/1/2017


{kind=link}
{kind=link}
{kind=link}