










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMedicina Interna
versão impressa ISSN 0872-671X
Medicina Interna vol.27 no.3 Lisboa jul. 2020
https://doi.org/10.24950/CC/71/20/3/2020
CASOS CLÍNICOS / CASE REPORTS
Neuromiotonia: Recordando uma Doença Rara
Neuromyotonia: Remembering a Rare Pathology
Sara Raquel Martins1  https://orcid.org/0000-0003-0978-5724
https://orcid.org/0000-0003-0978-5724
Sara Xavier Pires1 https://orcid.org/0000-0001-9760-2371
António Marinho2 https://orcid.org/0000-0002-3295-6723
1Departamento de Medicina Interna, Hospital de Santo António, Centro Hospitalar Universitário do Porto, Porto, Portugal
2Unidade de Imunologia Clínica, Hospital de Santo António, Centro Hospitalar Universitário do Porto, Porto, Portugal
Resumo:
A neuromiotonia é uma patologia muito rara, maioritariamente adquirida e de provável fundo autoimune. Caracteriza-se por hiperexcitabilidade dos nervos periféricos com subsequente hiperatividade e défice de relaxamento muscular; fazendo-se frequentemente acompanhar de distúrbios autonómicos e sensitivos. O diagnóstico estabelece-se através de eletromiografia, com o achado de múltiplos disparos espontâneos de potenciais de elevada frequência originados numa unidade motora única; sendo relevante não só para o controlo de sintomas potencialmente disruptivos com terapêutica adequada, mas também pela conhecida associação desta patologia a neoplasias – que devem sempre ser excluídas. No entanto, devido à raridade e à sintomatologia inespecífica e variável, o diagnóstico é difícil, sendo facilmente confundido com as mais comuns doenças psiquiátricas ou do primeiro neurónio, cujas terapêuticas podem resultar em iatrogenia adicional. Nesse sentido, trazemos um caso típico de neuromiotonia, a propósito da qual se recordam a sintomatologia habitual, meios complementares de diagnóstico, evolução natural e tratamentos efetivos da doença.
Palavras-chave: Síndrome de Isaac/diagnóstico; Síndrome de Isaac/tratamento.
Abstract:
Neuromyotonia is an exceedingly rare disease. In most cases it seems to be acquired, with a probable autoimmune origin. It is characterized by peripheral nerve hyperexcitability with muscle hyperactivity and deficient relaxation, and it is frequently accompanied by autonomic and sensitive symptoms. The diagnosis is established by electromyography, showing spontaneous, multiple single motor unit discharges, firing at a high frequency. The diagnosis is relevant not only to achieve symptom control but also for its known association with neoplasia, which must be ruled out. Nevertheless, the diagnosis is difficult to establish due to the nonspecific and variable symptoms that may lead to confusion with the more common psychiatric or motor neuron diseases. Misdiagnose can result in incorrect therapeutic actions with further iatrogenic symptoms. We report a typical case of neuromyotonia, in respect of which we review the clinical presentation, complementary diagnostic methods, natural evolution and treatment of the disease.
Keywords: Isaacs Syndrome/diagnosis; Isaacs Syndrome/ therapy.
Introdução
A neuromiotonia (ou síndrome de Isaac) é uma patologia muito rara, caracterizada por hiperexcitabilidade dos nervos periféricos com subsequente hiperatividade e défice de relaxamento muscular, cursando também com distúrbios autonómicos e sensitivos.1,2 Fisiopatologicamente parece ser uma doença de origem autoimune, com cerca de 40% dos pacientes a mostrarem anticorpos contra canais de potássio voltagem-dependentes.1 A eletromiografia (EMG) com deteção de disparos espontâneos de potenciais de elevada frequência originados numa unidade motora única é o goldstandard para diagnóstico.1-3 O diagnóstico é difícil pela clínica inespecífica e pela raridade da síndrome, no entanto relevante pela sintomatologia disruptiva que pode ser controlável com medicação, assim como pela possibilidade de associação a neoplasias que devem sempre ser excluídas.1,2
Caso clínico
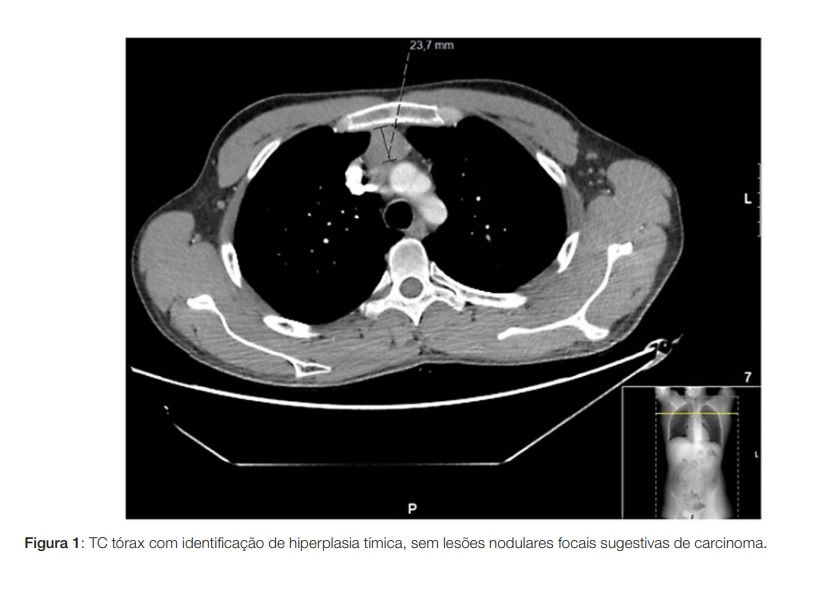
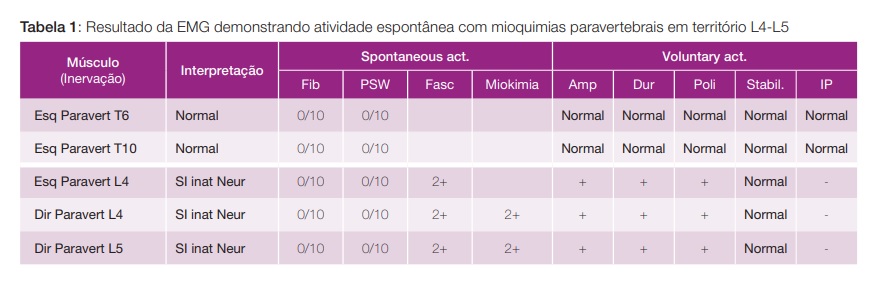
Apresentamos o caso de um homem de 29 anos, sem antecedentes pessoais ou história familiar relevantes, que se apresenta com quadro com um ano de evolução de mioquimias/movimentos involuntários dos membros inferiores e periorais, mialgias e fraqueza muscular de predomínio proximal, associados a lentificação psicomotora na realização de tarefas quotidianas. Concomitantemente com disfunção autonómica (hipersudorese, hipotensão ortostática, disfunção erétil e sialorreia), alterações sensitivas (disestesias em peúga), assim como disfagia para líquidos e perda ponderal de 10%. Ao exame objetivo apresentava força muscular conservada, mas fasciculações e mioquimias difusas de predomínio nos membros inferiores e língua; e hiperreflexia com hipertonia. Por ter sido previamente atribuída etiologia psiquiátrica à sintomatologia e instituída terapêutica com antipsicóticos mostrava ainda rigidez e bradicinésia bilaterais, resolvidas após suspensão dos fármacos. Sem alterações em estudo analítico extenso (incluindo serologias víricas, ceruloplasmina e estudo vitamínico) ou estudo imunológico (que incluiu anticorpos anticanais de potássio voltagem-dependentes e antirecetor pós-ganglionar de acetilcolina; anticorpos antigangliosídieo, anticorpos antineuronais e anticorpos anti-GAM (glicoproteína associada a mielina); assim como estudo de vasculites, lúpus, síndrome antifosfolipídica, síndrome de Sjögren e doença celíaca. Estudo de líquido cefalorraquidiano sem alterações. Ressonância magnética do neuroeixo e dos membros inferiores sem lesões. Tomografia computorizada (TC) cranioencefálica normal, realizou também TC toraco-abdomino-pélvico para avaliação de eventuais neoplasias que identificou apenas hiperplasia tímica (Fig. 1). Tomografia por emissão de positrões (PET) sem alterações hipermetabólicas sugestivas de patologia tumoral. EMG com “atividade elétrica muscular involuntária, em repouso, caraterizada por dupletos/tripletos e descargas mioquímicas circunscritas à musculatura paravertebral lombar, traduzindo uma hiperexcitabilidade do nervo periférico” (Tabela 1). Foi estabelecido assim o diagnóstico de neuromiotomia e instituída terapêutica desde logo com corticoterapia (bólus de 1 g de metilprednisolona durante 3 dias, seguido de prednisolona 30 mg/ dia), com excelente resposta. Posteriormente iniciada azatioprina 150 mg que permitiu o desmame lento e interrupção de corticóides sem recidiva da sintomatologia. Mantém vigilância em consulta externa (follow-up de 2 anos à data) e realização de TC toraco-abdomino-pélvico anual, apresentando o timo al guma involução das dimensões, sem identificação de novas lesões suspeitas. Tem vindo a tolerar desmame lento de azatioprina, atualmente medicado com 75 mg/dia.
Discussão
A neuromiotonia é uma patologia muito rara (prevalência de <1/1 000 000), caracterizada por hiperexcitabilidade do nervo periférico, que se traduz em hiperatividade muscular involuntária e contínua com clínica de cãibras, mioquimias, fasciculações, rigidez muscular e deficiente relaxamento muscular (pseudomiotonia).1,4 As mioquimias (contrações musculares rápidas, involuntárias) são o sintoma mais frequente (presente em 90% dos doentes) e geralmente são mais proeminentes nos membros inferiores, tronco e face, embora possam atingir outras regiões corporais.1 Disartria e disfagia podem surgir quando há atingimento de músculos bulbares.3 São comuns queixas concomitantes de distúrbios autonómicos (em particular hipersudorese, mas também alterações de foro gastrointestinal ou da regulação térmica, taquicardia ou taquipneia, disfunção erétil, entre outros) e mais raramente podem coexistir também alteraçõessensitivas (parestesias ou dor), assim como queixas de fraqueza muscular e fadiga, estando também reportada perda ponderal significativa.2 Alguns pacientes apresentam ainda alterações do sistema nervoso central (insónia, alterações cognitivas ou neuropsiquiátricas), sendo que a presença de encefalopatia passa a caracterizar a doença como síndrome de Morvan.3-5
Embora pontualmente possa surgir associada a doenças genéticas hereditárias, a neuromiotonia parece ser uma patologia maioritariamente adquirida.1,3 Nestes casos pressupõe-se uma etiologia autoimune pela constatação de resposta a imunoterapia, além da verificação de presença de bandas oligoclonais no líquido cefalorraquidiano de alguns pacientes.3 Para além disso, cerca de 40% dos pacientes com neuromiotomia adquirida apresentam anticorpos contra canais de potássio voltagem-dependentes, chegando esta associação a 80% nos casos secundários a timoma; e outros anticorpos (anticorpos antirecetor pós-ganglionar de acetilcolina) ou doenças de base imunitária (como miastenia gravis ou síndromes paraneoplásicos) podem ser detetados em 50% destes doentes.1,2,6,7
O diagnóstico exige deteção de atividade muscular contínua na eletromiografia, caracterizada por disparos espontâneos de potenciais de unidade motora em douplet, triplet ou múltiplos, com elevada frequência (30-300Hz). Atendendo à origem periférica da hiperexcitabilidade este padrão de descargas pode ser abolido apenas através de bloqueio neuromuscular; mantendo-se durante o sono, anestesia geral ou bloqueio de nervo periférico (contrariamente aos distúrbios de origem central). A presença de anticorpos anticanais de potássio voltagem-dependentes é condição facilitadora, mas não essencial, para o diagnóstico.1,2 Atendendo à conhecida descrição de neuromiotonia associada a neoplasias (timomas, linfomas, carcinoma de pequenas células do pulmão, neoplasias da próstata), estas devem ser ativamente pesquisadas em todos os doentes.1,7
Não obstante as associações acima referidas da neuromiotonia com timomas ou miastenia gravis, não se encontram registos de relação desta patologia com hiperplasia tímica apenas, pelo que se considerou que esta não justificaria o quadro clínico atual.
O controlo sintomático pode ser conseguido com terapêutica anticonvulsivantes (fenitoína, carbamezepina, valproato de sódio ou lamotroigina) mas frequentemente os doentes com neuromiotomia adquirida requerem imunossupressão (prednisolona e azatioprina) embora nem todos tenham uma resposta completa mesmo sob esta. A plasmaferese e imunoglobulinas podem conseguir um controlo transitório das sintomatologias graves.1,2,8,9
No presente caso optou-se por instituir imunossupressão ab initio devido à frequente falência dos anticonvulsivantes nos casos de neuromiotonia adquirida,1,2 ao facto de o doente já ter sido previamente medicado com estabilizadores de humor e neurolépticos tendo como resultado agravamento do quadro, e ainda atendendo ao sentido fisiopatológico da imunossupressão dado a pressuposta base imunológica desta patologia.
A história natural da doença é variável, por vezes cursando com remissão completa da clínica em alguns meses enquanto que noutros pacientes a patologia pode persistir durante vários anos. Nos doentes com sintomatologia refratária a imunoterapias devem ser ponderadas etiologias não-imunológicas.7
Conclusão
A neuromiotonia é uma patologia muito rara, cuja versatilidade de apresentações facilmente induz diagnósticos erróneos. Assim sendo, a sensibilidade clínica é essencial para a sua identificação, a par com os achados característicos da eletromiografia. Pode causar sintomatologia disruptiva, mas controlável com tratamento, o qual passa por anticonvulsivantes e/ou imunossupressão. A associação com neoplasias torna obrigatória a sua exclusão em todos os casos de neuromiotonia adquirida.
REFERÊNCIAS
1. Maddison P. Neuromyotonia. Pract Neurol. 2002;2:225-9. doi: 10.1046/j.1474-7766.2002.00068.x [ Links ]
2. Ahmed A, Simmons Z. Isaacs syndrome: A review. Muscle Nerve. 2015;52:5-12. doi: 10.1002/mus.24632 [ Links ]
3. Sawlani K, Katirji B. Peripheral nerve hyperexcitability syndromes. Continuum. 2017;23:1437–50. doi: 10.1212/CON.0000000000000520
4. Orpha.net [homepage na Internet]. Paris: Instituto Nacional Francês para a Saúde e Investigação Médica. [consultado 10 Abril 2020] Disponível em: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=84142
5. Lee W, Day TJ, Williams DR.Clinical, laboratory and electrophysiological features of Morvan’s Fibrillary Chorea. J Clin Neurosci. 2013;20:1246–9. doi: 10.1016/j.jocn.2012.10.029
6. Hart IK, Maddison P, Newsome-Davis J, Vincent A, Mills KR. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain. 2002;125:1887–95. doi: 10.1093/brain/awf178
7. Vincent A., Pettingill P., Pettingill R., Lang B., Birch R.,Waters P, et al. Association of leucine-rich glioma inactivated protein 1, contactin associated protein 2, and contactin 2 antibodies with clinical features and patient-reported pain in acquired neuromyotonia. JAMA Neurol. 2018;75:1519–27. doi: 10.1001/jamaneurol.2018.2681
8. Nakatsuji Y, Kaido M, Sugai F, Nakamori M, Abe K, Watanabe O, Arimura K, Sakoda S. Isaacs' syndrome successfully treated by immunoadsorption plasmapheresis. Acta Neurol Scand. 2000;102:271-3. doi: 10.1034/j.1600-0404.2000.102004271.x. [ Links ]
9. Béquet D, Devière F, Renard JL, Felten D. Syndrome d'Isaacs: amélioration durable par les immunoglobulines polyvalentes intraveineuses. Rev Neurol. 1997;153:602-4. [ Links ]
Responsabilidades Éticas
Conflitos de Interesse: Os autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
Fontes de Financiamento: Não existiram fontes externas de financiamento para a realização deste artigo.
Confidencialidade dos Dados: Os autores declaram ter seguido os protocolos da sua instituição acerca da publicação dos dados de doentes.
Consentimento: Consentimento do doente para publicação obtido. Proveniência e Revisão por Pares: Não comissionado; revisão externa por pares.
Ethical Disclosures
Conflicts of interest: The authors have no conflicts of interest to declare.
Financing Support: This work has not received any contribution, grant or scholarship
Confidentiality of Data: The authors declare that they have followed the protocols of their work center on the publication of data from patients.
Patient Consent: Consent for publication was obtained.
Provenance and Peer Review: Not commissioned; externally peer reviewed.
© Autor (es) (ou seu (s) empregador (es)) e Revista SPMI 2020. Reutilização permitida de acordo com CC BY-NC. Nenhuma reutilização comercial.
© Author(s) (or their employer(s)) and SPMI Journal 2020. Re-use permitted under CC BY-NC. No commercial re-use.
Correspondence / Correspondência:
Sara Raquel Martins – sararaquelm@gmail.com
Departamento de Medicina Interna, Hospital de Santo António, Centro Hospitalar Universitário do Porto, Porto, Portugal
Largo Prof. Abel Salazar 4099-001 PORTO
Received / Recebido: 22/04/2020
Accepted / Aceite: 07/06/2020
Publicado / Published: 28 de Setembro de 2020


{kind=link}
{kind=link}