










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMedicina Interna
versão impressa ISSN 0872-671X
Medicina Interna vol.27 no.4 Lisboa dez. 2020
https://doi.org/10.24950/CC/93/20/4/2020
CASOS CLÍNICOS / CASE REPORTS
Isolated Nodal Retroperitoneal Sporadic Lymphangioleiomyomatosis
Linfangioleiomiomatose Esporádica Ganglionar Retroperitoneal Isolada
Bárbara Soeiro1  https://orcid.org/0000-0001-9545-6534
https://orcid.org/0000-0001-9545-6534
João Correia-Pinto2 https://orcid.org/0000-0003-1993-1705
Alexandre Vasconcelos1
Matilde Salgado3
1Serviço de Medicina, Unidade Local de Saúde de Matosinhos, Hospital Pedro Hispano, Matosinhos, Portugal
2Serviço de Anatomia Patológica, Unidade Local de Saúde de Matosinhos, Hospital Pedro Hispano, Matosinhos, Portugal
3Serviço de Oncologia, Unidade Local de Saúde de Matosinhos, Hospital Pedro Hispano, Matosinhos, Portugal
Abstract:
Lymphangioleiomyomatosis is a rare disease (1/1 000000) with multisystemic involvement, most commonly presenting with cystic lung disease. Reports of isolated nodal lymphangioleiomyomatosis rare. Its treatment and prognosis are not well established. We describe a 48-year-old female patient presenting with ureter compression by an extensive retroperitoneal mass. Biopsy showed nodal lymphangioleiomiomatosis. A partial surgical resection of the lesions was performed. She is under yearly thoraco-abdomino-pelvic computed tomography control, last performed 3 years after diagnosis, with no lung involvement or new abdominal lesions. She evolved to menopause after 6 months. Some case series suggest that isolated nodal lymphangioleiomyomatosis may precede lung involvement by one to two years, having size as a risk factor (>10 mm). Relative stabilization of the disease course in post-menopausal women is described. This case reports an atypical evolution, with two >10mm diameter lesion, with3-year follow-up without pulmonary, renal or other forms of lymphatic involvement.
Keywords: Lymphangioleiomyomatosis; Lymph Nodes; Menopause.
Resumo:
A linfangioleiomiomatose é uma doença rara multissitémica (1/1 000 000), apresentado-se tipicamente com doença pulmonar cística. Relatos de linfangioleiomiomatose ganglionar isolada são muito raros. Existe pouca informação relativamente ao tratamento e prognóstico. Descrevemos o caso de uma mulher de 48 anos com compressão uretérica por uma massa retroperitoneal. A biópsia mostrou linfangioleiomiomatose ganglionar. Foi submetida a ressecção parcial e mantém-se sob vigilância imagiológica há 3 anos, sem evidência de envolvimento pulmonar ou aumento das lesões abdominais. A doente evoluiu para menopausa 6 meses após o diagnóstico. Séries de casos sugerem que a linfangioleiomiomatose ganglionar pode preceder o envolvimento pulmonar em 1-2 anos, sendo as dimensões (>10 mm) da lesão um fator de risco. Está descrita estabilização da doença em mulheres pós-menopausa. Este caso evidencia uma apresentação e evolução atípicas, com uma lesão com >10 mm, com 3 anos de follow-up sem atingimento pulmonar, renal ou outras formas de atingimento linfático mais frequentes.
Palavras-chave: Linfangioleiomiomatose; Menopausa; Nódulos Linfáticos.
Introduction
Lymphangioleiomyomatosis is a rare disease (1/1 00000) with multisystemic involvement, that occurs mainly in female individuals in the 4th decade of life. It is usually associated with tuberous sclerosis complex (TSC). When isolated it is called sporadic lymphangioleiomyomatosis (LAM) and lacks the cutaneous manifestations (e.g. fibromas, facial angiomas, hypomelanotic macules). The primary affected organ of sporadic LAM is the lung (bilateral thin-walled cysts in chest-computed tomography (CT)). Extra-pulmonary LAM is rarer, involvinglesions of the lymphatic vasculature (chylothorax, chylous ascites and lymph node lymphangioleiomyomatosis) and renal angiomyolipomas (AMLs). Lymphadenopathy, especially retroperitoneal and pelvic, is not uncommon in patients with pulmonary LAM, but no study has specifically examined the prognostic significance of small, incidental LAM occasionally identified in lymph nodes resected for unrelated purposes.1-5
Case Report
We report the case of a 48-year-old Caucasian female patient, with history of arterial hypertension and dyslipidaemia and renal lithiasis treated with lithotripsy 7 years before our observation. She hadpast resection of intra-abdominal and umbilical endometriosis lesions and elective fallopian tube occlusion at the age of 37 years old. She had history of one caesarean delivery and one-yearlong peri-menopause phase. She was not under any exogenous systemic estrogen therapy.
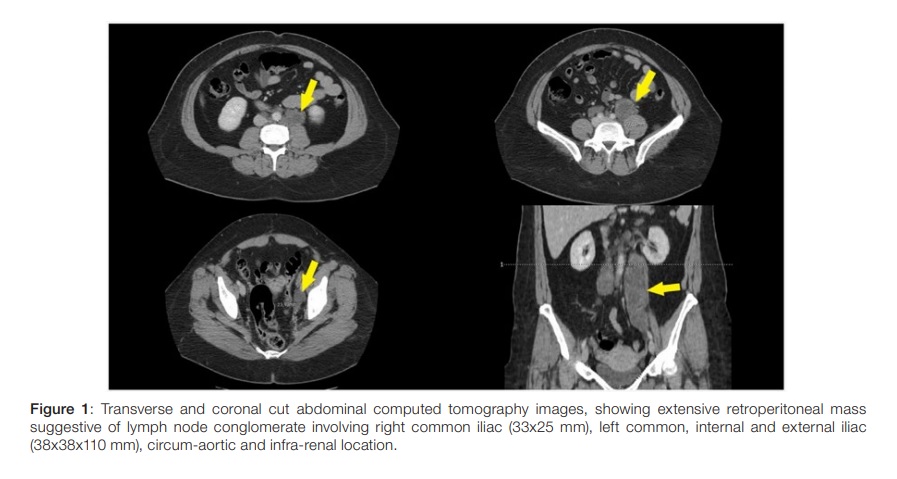
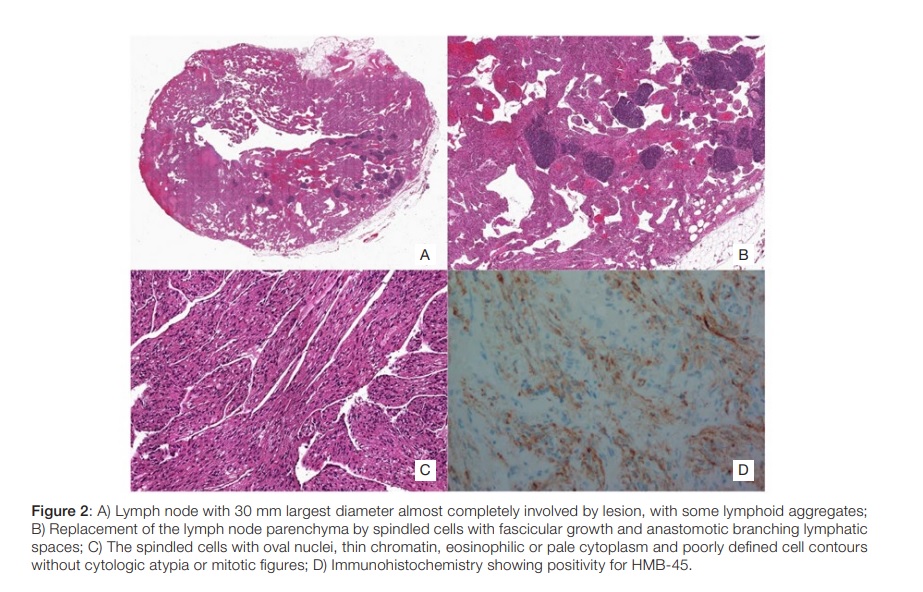
She presented with one-week evolution of left low back pain, progressing to 7 in 10 intensity. In the previous 3 months she noticed unspecific abdominal volume increase. She exhibited left renal murphy sign and moderate discomfort on left iliac fossa palpation,withno peritoneal irritation signs. She presented microscopic haematuria (43 erythrocytes per high-power field) and left hydronephrosis (10 mm of transverse diameter on proximal ureter) on immediate renal echography, with no visible calculus. Abdominopelvic CT revealed extensive retroperitoneal mass suggestive of lymph node conglomerate involving right common iliac (33x25 mm), left common, internal and external iliac (38x38x110 mm), circum-aortic and infra-renal location, causing left ureter compression (Fig. 1). At this point, the differential diagnosis comprehended retroperitoneal fibrosis, primary or metastatic solid tumors or lymphoproliferative neoplasm. Laparoscopic biopsy was performed. Histology showed replacement of the lymph node parenchyma by spindled cells with fascicular growth and anastomotic branching lymphatic spaces. The spindled cells presented oval nuclei, thin chromatin, eosinophilic or pale cytoplasm and poorly defined cell contours without cytologic atypia or mitotic figures. Immunohistochemistry was performed and showed expression of smooth-muscle actin, desmin, HMB-45 and estrogen receptors and it was negative for Melan-A, compatible with nodal lymphangioleiomiomatosis (Fig. 2).
Chest-CT displayed no lung involvement and abdominal CT no renal angiomyolipomas.
Due to the ureteric compression, an elective surgical resection of the lesions was performed with no complications, but no complete excision. She was started on tamoxifen 20 mg a day, with good tolerance. She evolved to menopause around 6 months after diagnosis. She is under yearly thoraco-abdomino-pelvic CT control, last performed 3 years after diagnosis, with no lung involvement, no new abdominal / retroperitoneal lesions and dimensional stability of non-resected lesions.
Discussion
The diagnosis of nodal LAM is usually straightforward. Most cases can be identified by hematoxylin and eosin staining due to the particularmorphology, but insome settings immunohistochemical tests can behelpful. The gold-standard immunohistochemical diagnostic for LAM is positive immunostaining withHMB-45 antibody.5 Besides HMB-45, the most commonly used markers in the immunohistochemical diagnosis of LAM are á-smooth muscle actin (SMA), estrogen receptor (ER), and progesterone receptor (PR).6 Schoolmeester and colleagues6 examined the expression of HMB45, â-catenin and Melan-A in 18 cases of nodal LAM and found that HMB45 and â-catenin were the more consistently positive markersin nodal LAM (100% of cases) than Melan-A (39%).
Reports of isolated retroperitoneal lymphangioleiomyomatosis are very rare in the literature. As so, its treatment and prognosis are not well established. A study of 22 patients with nodal LAM, foundthat a diagnosis ofnodal LAM preceded development of pulmonary LAM by 1 to 2 years. 11 patients remained asymptomatic and 9 patients hadsignsor symp- toms related solely to abdominopelvic nodal LAM. A relation between size of the affected node (atleast one node with more than 10 mm) and subsequent or simultaneous development of pulmonary LAM was suggested.1 Schoolmeester and colleagues2 analysed 19 patients with lymph node (pelvic and retroperitoneal) LAM, none had a history of TSC, renal AML or pulmonary LAM. Eighteen patients had surgery for tumours involving the reproductive system and ganglionar involvement was an incidental finding. All 18 patients without recurrent or persistent LAM were under 10mm in greatest dimension.
The difficulty in developing useful therapeutic options for LAM is that the true source of the LAM cell is unknown and the mechanisms that contribute to LAM cell proliferation, metastasis and migration are not fully understood. Positive results are shown on the stabilization of pulmonary function, a reduction of angiomyolipoma size,and rare reports of affected abdominal and pelvic lymph node size reduction with inhibitors of the mammalian target of rapamycin (mTOR). Sirolimus and everolimus are recommended in pulmonary LAM in the Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines.7 The benefits on extra-pulmonary LAM are not well established.8-10 Estrogen and progesterone receptors are thought to play arole on tumor proliferation. Contradictory results with hormonal therapy, mainly in pulmonary LAM, have been reported in case series.11 The Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines do not recommend its use.7 Inhibition of aromatase to block estrogen synthesis is also conflicting.12 Relative stabilization of the disease course in postmenopausal women with LAM has been described13,14 and may have played an important role in this case. This case reports an atypical evolution, with two >10mm diameter lesion, with three-year follow-up without pulmonary, renalor other more frequent formsof lymphatic involvement.
REFERENCES
1. Matsui K, Tatsuguchi A, Valencia J, Yu Zx, Bechtle J, Beasley MB, et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol. 2000;31:1242. doi: 10.1053/hupa.2000.18500. [ Links ]
2. Schoolmeester JK, Park KJ. Incidental Nodal Lymphangioleiomyomatosis Is Not a Harbinger of Pulmonary Lymphangioleiomyomatosis: A Study of 19 Cases with Evaluation of Diagnostic Immunohistochemistry. Am JSurg Pathol. 2015;39:1404-10. doi: 10.1097/PAS.0000000000000470. [ Links ]
3. Tobino K, Johkoh T, Fujimoto K, Sakai F, Arakawa H, Kurihara M, et al. Computed tomographic features of lymphangioleiomyomatosis: evalua tion in 138 patients. Eur J Radiol. 2015 Mar;84(3):534-41. doi: 10.1016/j.ejrad.2014.12.008. [ Links ]
4. Urban T, Lazor R, Lacronique J, Murris M, Labrune S, Valeyre D, et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur lesMaladies "Orphelines" Pulmonaires (GERM"O"P). Medicine. 1999;78:32137. DOI: 10.1097/00005792-199909000-00004.
5. Johnson SR, Cordier JF, Lazor R, Cottin V, Costabel U, Harari S, et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010; 35: 14-26. doi: 10.1183/09031936.00076209. [ Links ]
6. Grzegorek I, Drozdz K, Podhorska-Okolow M, Szuba A, Dziegiel P, et al. LAM cells: biology and Lymphangioleiomyomatosis. Folia Histochem Cytobiol. 2013; 51: 1-10. doi: 10.5603/FHC.2013.001. [ Links ]
7. McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, et al. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am J Respir Crit Care Med.2016;194:748–61. doi: 10.1164/rccm.201607-1384ST.
8. Wahid S, Chiang PC, Lun Luo H, Huan SC, Tsai EM, Chiang PH. Pelvic lymphangioleiomyomatosis treated successfully with everolimus - Two case reports with literature review. Medicine. 2017; 96: e4562. doi: 10.1097/MD.0000000000004562. [ Links ]
9. Freitas C, Baldi B, Araújo M, Heiden GI, Kairalla RA, Carvalho CR. Use of sirolimus in the treatment of lymphangioleiomyomatosis: favorable responses in patients with different extrapulmonary manifestations. J. Bras Pneumol. 2015;41:275-80. Doi: 10.1590/S1806-37132015000004553. [ Links ]
10. Cai Y, Guo H, Wang W, Li H, Sun H, Shi B, et al. Assessing the outcomes of everolimus on renal angiomyolipoma associated with tuberous sclerosis complex in China: a two years trial. Orphanet J Rare Dis. 2018;13:43. doi: 10.1186/s13023-018-0781-y. [ Links ]
11. Schiavina M, Contini P, Fabiani A, Cinelli F, Di Scioscio V, Zompatori M, et al. Efficacy of hormonal manipulation in lymphangioleiomyomatosis. A 20-year-experience in 36 patients. Sarcoidosis Vasc Diffuse Lung Dis. 2007;24:39-50. [ Links ]
12. Taveira-DaSilva AM, Moss J. Management of lymphangioleiomyomatosis. F1000Prime Rep. 2014;6:116. doi: 10.12703/P6-116. [ Links ]
13. Johnson SR, Tattersfield AE. Decline in lungfunction in lymphangioleiomyomatosis: relation to menopause and progesterone treatment. Am J Respir Crit Care Med. 1999;160:628–33. doi: 10.1164/ajrccm.160.2.9901027.
14. Gupta N, Lee HS, Young LR, Strange C, Moss J, Singer LG, et al. Analysis of the MILES cohort reveals determinants of disease progression and treatment response in lymphangioleiomyomatosis. Eur Respir J. 2019;53:1802066. doi: 10.1183/13993003.02066-2018. [ Links ]
Ethical Disclosures
Conflicts of interest: The authors have no conflicts of interest to declare.
Financing Support: This work has not received any contribution, grant or scholarship
Confidentiality of Data: The authors declare that they have followed the protocols of their work center on the publication of data from patients.
Patient Consent: Consent for publication was obtained.
Provenance and Peer Review: Not commissioned; externally peer reviewed.
Responsabilidades Éticas
Conflitos de Interesse: Os autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
Fontes de Financiamento: Não existiram fontes externas de financiamento para a realização deste artigo.
Confidencialidade dos Dados: Os autores declaram ter seguido os protocolos da sua instituição acerca da publicação dos dados de doentes.
Consentimento: Consentimento do doente para publicação obtido.
Proveniência e Revisão por Pares: Não comissionado; revisão externa por pares.
© Author(s) (or their employer(s)) and SPMI Journal 2020. Re-use permitted under CC BY-NC. No commercial re-use.
© Autor (es) (ou seu (s) empregador (es)) e Revista SPMI 2020. Reutilização permitida de acordo com BY-NC. Nenhuma reutilização comercial.
Correspondence / Correspondência:
Bárbara Soeiro – barbarasoeiro@hotmail.com
Serviço de Medicina, Unidade Local de Saúde de Matosinhos, Hospital Pedro Hispano, Matosinhos, Portugal
Morada: Rua Dr. Eduardo Torres, 4464-513 Senhora da Hora
Received / Recebido: 20/05/2020
Accepted / Aceite: 05/07/2020
Publicado / Published: 18 de Dezembro de 2020


{kind=link}
{kind=link}