











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
Valores de triglicéridos (TG) (Tabela 1) superiores a 150 mg/dL conferem um aumento do risco vascular com base em estudos observacionais de larga dimensão, não estan-do o mecanismo causal na doença cardiovascular comple-tamente esclarecido.1,2Além do risco cardiovascular, níveis muito, elevados de TG associam-se a pancreatite.
Tabela 1: Lista de acrónimos
| APO A5: Apolipoproteína A5 |
| Apo B: Apolipoproteína B |
| Apo C II: Apolipoproteína C II |
| Apo C III: Apoproteína C III |
| Col: Colesterol |
| γGT: gamaglutamil transferase |
| GPIHBP1: Proteína de ligação à lipoproteína de alta densidade ancorada ao glicosilfosfatidilinositol |
| HDL: Lipoproteína de alta densidade |
| IMC: índice de massa corporal |
| LDL: Lipoproteína de baixa densidade |
| LMF1: Fator de maturação da lipase 1LPL: Lipoproteína lipase |
| Lp(a): Lipoproteína (a) |
| QM: Quilomicron |
| QMM: Quilomicronémia multifactorial |
| SQF: Síndrome da quilomicronémia familiar |
| TG; Triglicéridos |
| VLDL- very low density lipoprotein |
Definem-se níveis normais de triglicéridos se forem in-feriores a 150 mg/dL, hipertrigliceridemia moderada entre 150 e 499 mg/dL e grave quando igual ou superior a 500 mg/dL.3 Adotamos a definição de quilomicronemia quando os valores de triglicéridos são superiores a 880 mg/dL (10 mmol/L); esta situação representa um excesso de quilomicra na circulação sanguínea, lipoproteínas ricas em TG produzidas pelos enterócitos após uma refeição.1,4
Em Portugal, a hipertrigliceridemia tem uma prevalência estimada de 13%. Contudo, a hipertrigliceridemia grave não se encontra devidamente estudada, podendo apenas inferir-se que estará presente em menos de 0,7% da população, afetando sobretudo os homens (1,2% vs 0,2%: de acordo com o estudo VALSIM).5
Dados extrapolados do estudo NHANES apontam para que 1,7% da população adulta dos Estados Unidos tenha ní-veis de TG entre 500 e 2000 mg/dL.2 No Copenhagen Gene-ral Population Study, a frequência da hipertrigliceridemia entre 442 e 885 mg/dL foi de 1% nas mulheres e 3% em homens; a quilomicronemia (triglicéridos superiores a 885 mg/dL) registou-se em 0,03% das mulheres e 0,14% dos homens.6
A quilomicronemia pode ser diferenciada em dois subtipos, clinicamente semelhantes, mas com diferentes etiologias, e consequentemente com respostas diferentes à terapêutica: síndrome de quilomicronemia familiar (SQF) e quilomicronemia multifactorial (QMM).7
FISIOPATOLOGIA DA QUILOMICRONEMIA
Os quilomicra são habitualmente removidos do plasma pela ação da lipoproteína lipase (LPL), enzima presente na superfície das células adiposas e musculares, que hidrolisa os QM a remanescentes, sendo depois captados pelas células. A redução da actividade da LPL pode dever-se a mutações do gene da LPL ou de outras proteínas envolvidas como seus cofactores.8
O metabolismo dos triglicéridos é controlado por múltiplas proteínas das quais duas têm um papel fundamental: apoC-III e ANGPTL3. A apoC-III é uma glicoproteína (sintetizada sobretudo no fígado, mas também no intestino) que se liga à superfície de quase todas as lipoproteínas. Estão propostos 2 mecanismos pelos quais a apoC-III aumenta os TG: inibição da lipólise mediada pela LPL e/ou atenuação da captação hepática dos TG, aparentando ser este último o predominante no SQF.9
Além dos seus efeitos no metabolismo das lipoproteínas, a apoC-III tem também propriedades pro-ateroscleróticas intrínsecas: ela promove a adesão dos monócitos ao endotélio, aumenta a produção de mediadores inflamatórios e aumenta a retenção de LDL na parede arterial.10
A ANGPTL3 é uma proteína produzida no fígado, também inibidora da LPL e da lipase endotelial.11 Mutações de perda de função na ANGPTL3 em homozigotia resultam numa redução significativa de todas as lipoproteínas plasmáticas excepto da Lp(a). Apesar de o número de homozigotos ser reduzido, parecem partilhar características como défice da lipemia pós-prandial, diabetes mellitus, esteatose hepática e o portadores.12
SÍNDROME DA QUILOMICRONÉMIA FAMILIAR OU QUILOMICRONEMIA MONONOGÉNICA
A síndrome da quilomicronemia familiar (SQF), quilomi-cronemia monogénica, tradicionalmente conhecida como hiperlipoproteinemia tipo 1 (OMIM#238600) é uma doença autossómica recessiva rara do metabolismo dos quilomicra. A SQF caracteriza-se por níveis de TG acima de 880 mg/dL na ausência de medidas terapêuticas.
Estima-se que a quilomicronemia possa ocorrer em 1:600 adultos, sendo o SQF uma parcela reduzida destes casos.7 A SQF tem uma hereditariedade autossómica recessiva, com uma prevalência estimada de cerca de 1 em cada 1 000 000 de habitantes.
São conhecidas mutações em diversos genes relacionados com a lipólise dos quilomicra, podendo todas estas mutações apresentar-se com o fenótipo de SQF:
1) Lipoproteína lipase (LPL), responsável por mais de 90%dos casos
2) Apolipoproteína C2 (APOC2);
3) Apolipoproteína A5 (APOA5);
4) Factor de maturação da lipase 1 (LMF1);
5) Proteína de ligação à lipoproteína de alta densidade an-corada ao glicosilfosfatidilinositol (GPIHBP1).
Mutações destes genes em homozigotia ou heterozigotia composta, comprometem a clearance dos quilomicra, com valores de TG doseados em jejum superiores a 900 mg/dL.7 A tendência verificada é de valores mais elevados de TG nos doentes com mutação da LPL do que nos restantes genes que referimos.13
Classicamente é descrito que o plasma destes pacien-tes tem um aspeto leitoso, visualizando-se um sobrenadante composto por quilomicra após decantação ou centrifugação do plasma.10
Os valores de triglicéridos em doentes com SQF são habitualmente 10 vezes mais elevados que o limite superior do normal, embora possam estar elevados mais de 100 vezes.14
Apesar de os factores ambientais intensificarem a quilomicronemia, não são necessários para a expressão da doença.8
Em relação ao risco cardiovascular e aterogénese, os valores de colesterol total são igualmente elevados nos doentes com mutação da LPL e dos restantes genes, ao passo que o colesterol LDL, colesterol HDL, ApoB e VLDL são baixos em todos os doentes com SQF. Este facto salienta uma caracte-rística metabólica fundamental ao SQF: dada a ausência ou mínima actividade da LPL, há um défice de conversão das partículas ricas em TG a formas remanescentes.13
MANIFESTAÇÕES CLÍNICAS DA SQF
Os portadores possuem elevadas concentrações de quilomicra em jejum, mas habitualmente não desenvolvem aterosclerose prematura, provavelmente pelo tamanho dos quilomicra, que impede as partículas de atravessar a barreira endotelial.1
As manifestações clínicas são múltiplas, destacando-se a dor abdominal (relacionada ou não com a pancreatite) e a pancreatite recorrente, que pode ser fatal, ou evoluir para pancreatite crónica. Esta condição prende-se com elevada morbilidade devido à insuficiência pancreática e diabetes tipo 3 associadas. Nas manifestações da SQF contam-se ainda os xantomas eruptivos do tronco e membros (em menos de 50% dos doentes), lipemia retinalis (aspecto leitoso dos vasos da retina), hepatoesplenomegalia, atraso no desenvolvimento, náuseas, vómitos, artralgias, fadiga e sintomas neurológicos (irritabilidade, défice de memória, demência, depressão). A qualidade de vida e a capacidade para o trabalho podem ser gravemente afetadas.8,14-16
A hipertrigliceridemia é a terceira causa mais frequente de pancreatite, seguindo-se às etiologias alcoólica e litiásica. Uma revisão sistemática identificou a hipertrigliceridemia grave como causa de 9% das pancreatites agudas.17
A pancreatite aguda é a complicação mais grave da hipertrigliceridemia. Pode ser fatal, e se recorrente, originar pancreatite crónica com insuficiência endócrina e exócrina.7 Está associada com uma evolução mais grave do que a pancreatite que decorre de outras etiologias, com maior incidência de falência de órgão, necrose pancreática e necessidade de cuidados intensivos.14 Não existe outro biomarcador identificado para predizer o risco de pancreatite. Assim, a redução da hipertrigliceridemia para menos de 885 mg/dL é o único alvo válido para a redução do risco.8
Uma análise interina ao estudo IN-FOCUS, um questionário online respondido por 60 doentes com SQF, permitiu destacar os sintomas múltiplos e heterogéneos experien-ciados por estes doentes.18 Entre 22% e 35% dos doentes referiram distensão ou dor abdominal, dispepsia, astenia ou fadiga; 42% tinham história de pancreatite aguda. A ameaça constante de um episódio de pancreatite aguda debilitante ou potencialmente fatal prejudicava gravemente o bem-estar do indivíduo em 1/3 dos casos. Mais de 90% dos doentes que integraram este estudo reportaram que o seu diagnóstico afetou as suas oportunidades e escolhas profissionais.20
As restrições alimentares ditadas pelas recomendações terapêuticas podem causar ansiedade relacionada com a alimentação e ter um impacto negativo na qualidade de vida.14 No IN-FOCUS, foi reportado que aplicar as restrições alimentares consumia muito tempo e se tornava esgotante (81% e 70% respetivamente), o que conduzia a ansiedade e isolamento social. Apesar destas restrições, 53% dos doentes mantinha-se sintomático.19
QUILOMICRONEMIA MULTIFACTORIAL (QMM)
A QMM, por vezes designada hiperlipoproteinemia tipo V é uma doença oligo/poligénica exacerbada pela presença de causas secundárias de hipertrigliceridemia (Tabela 2), surgindo habitualmente em idades mais avançadas que o SQF.7
Tabela 2: Causas secundárias de hipertrigliceridemia
| Gravidez | Fármacos |
|---|---|
| Obesidade | Anti-retrovirais (inibidores da protease) |
| Dieta com excesso calórico | Beta-bloqueantes não selectivos |
| Álcool | Glicocorticóides |
| Síndrome metabólico | Terapêutica de substituição hormonal |
| Diabetes mellitus | Tamoxifeno, raloxifeno |
| Hipotiroidismo | Tiazidas |
| Hipopituitarismo | Sequestradores de ácido biliares |
| Acromegália | Ciclofosfamida |
| Síndrome de Cushing | nnnnnnnnnnnnnnnnnnnnntipsicóticos de segunda geração (ex: clozapina, olanzapina, mirtazapina) |
| Lipodistrofia parcial familiar | Sirolimus, tacrolimus |
| Glicogenose | Propofol |
| Síndrome nefrótico | Interferão alfa |
| Paraproteinémia | L-asparaginase |
| Gamapatia monoclonal | |
| Lupus eritematoso sistémico | Adaptado de:1,3,6,10 |
Resulta de diversas variáveis predisponentes, que vão desde mutações de perda de função em heterozigotia, a polimorfismos funcionais, quando associados a comorbilidades ou factores ambientais classicamente associados a elevação dos triglicéridos.20,21
Num estudo que envolveu a caracterização genética de mais de 500 doentes com hipertrigliceridemia grave, 1,1 % eram homozigotos ou heterozigotos compostos para varian-tes genéticas raras (SQF), 14,4% apresentavam heterozigotia para variantes raras e em cerca de um terço (32%) identificou-se uma acumulação de várias alterações comuns (elevado score para hipertrigliceridemia poligénica).22
Salienta-se ainda que o alelo E2 da apolipoproteína E está presente na maioria dos doentes com QMM pelo que é prová-vel que contribua para a redução da clearance dos remanes-centes de quilomicra no fígado em doentes já com variantes patogénicas que alteram a actividade da LPL. Contudo a quilomicronemia não é comum mesmo em homozigóticos E2/E2 pelo que devem ser classificados como QMM.8
DIAGNÓSTICO DIFERENCIAL ENTRE SQF E QMM
A semelhança fenotípica entre o SQF e QMM em conjun-to com o desconhecimento da doença conduz ao subdiagnóstico da SQF, sendo frequente uma história de episódios recorrentes de pancreatite aguda antes do diagnóstico, com referenciação e seguimento inadequados.8
Embora com valores de triglicéridos elevados, os doentes com QMM têm história de pancreatite aguda com menos frequência que os doentes com SQF (6% vs 60%) assim como de episódios de dor abdominal ou pancreatite recorrente.7
Na QMM, a variabilidade dos níveis de TG é superior; a resposta à dieta e ao tratamento farmacológico com fibratos é significativa ao contrário da SQF.8
O diagnóstico diferencial entre SQF e QMM é auxiliado pela idade da primeira manifestação, o índice de massa cor-poral (IMC) e a gamaglutamil transferase. Os doentes com SQF têm manifestações clínicas mais precoces, IMC mais baixo e níveis de γGT inferiores que os doentes com QMM7 (Tabela 3).
Tabela 3: Principais diferenças clínicas entre SQF e QMM
| Síndrome quilomicronemia familiar (SQF) | Quilomicronemia multifactorial (QMM) | |
|---|---|---|
| Idade do diagnóstico | Mais jovem | Mais tardia |
| Dor abdominal e Pancreatite | Mais frequente, com início em idade jovem e recorrente | Menor frequência, mais tardia e recorrência menor |
| IMC | Em geral baixo | Habitualmente mais elevado |
| Níveis de triglicéridos | Níveis +++ em permanência | Níveis ++ e com variabilidade |
| Resposta à terapêutica convencional | Pouco eficaz | Eficaz |
| Risco cardiovascular | Menor | Maior |
Já foi sugerido que o risco cardiovascular é inferior nos doentes com SQF em comparação à QMM.23
A diferenciação entre SQF e QMM é possível com base na eletroforese das lipoproteínas ou na ultracentrifugação. No caso desta última, enquanto que no SQF apenas se demonstram quilomicra, na QMM são detetados quilomicra e VLDL.7 O lipidograma não é fiável dado que os quilomicra de maior dimensão com frequência não penetram no gel de agarose e não se detetam consistentemente.8
No passado, o teste de heparina foi importante no diagnóstico. A injeção intravenosa de heparina liberta a LPL do endotélio o que permite a determinação da sua actividade.14 O ensaio de actividade da LPL baseia-se na determinação da actividade da LPL que se mostra dramaticamente reduzida mesmo após injeção de 50 IU/kg de heparina.8 Níveis abaixo dos 20% são sugestivos de SQF.
Actualmente o teste genético é o gold standard diagnóstico.
TERAPÊUTICA DA SQF
O tratamento da hipertrigliceridemia tem dois objectivos distintos: a prevenção imediata da pancreatite em doentes com hipertrigliceridemia grave e a redução do risco cardio-vascular global. Uma vez que a hipertrigliceridemia se caracteriza por elevação dos quilomicra remanescentes, as concentrações do colesterol não-HDL ou ApoB são alvos secundários de tratamento.1
A base terapêutica da hipertrigliceridemia grave, consiste em dieta pobre em gorduras e hidratos de carbono e evicção de álcool.24 Os doentes com QMM respondem a medidas modificadoras do estilo de vida e correção das causas secundárias de hipertrigliceridemia assim como à terapêutica farmacológica.
Pelo contrário, os doentes com SQF respondem mal à terapêutica farmacológica pelo que o tratamento mais eficaz até recentemente tem sido a restrição dietética ex-trema.7 Uma dieta extremamente rigorosa com menos de 20 g de gordura por dia tem algum resultado nos doentes com SQF. Também a utilização de ácidos gordos de cadeia média tem resultados interessantes neste contexto e pode reduzir os níveis de triglicéridos para valores abaixo do limiar do risco de pancreatite. Ambas as estratégias são mal toleradas e difíceis de manter a longo prazo.6
Uma vez que a maioria das intervenções que reduzem os triglicéridos atuam, pelo menos parcialmente, pela clea-rance mediada pela LPL, estas não têm efeito na SQF.8 Agentes hipolipemiantes como estatinas, fibratos, ómega 3 e ácido nicotínico são ineficazes na SQF pois actuam na redução da síntese de VLDL no fígado e pelo incremento na actividade da LPL, o que requer a actividade desta enzima, que se encontra ausente ou reduzida no SQF.25
Apesar de estarem descritos casos isolados de plasma-ferese para controlo dos níveis de hipertrigliceridemia grave, esta técnica é utilizada geralmente apenas no contexto de pancreatite aguda, pelo que não será alvo de análise neste artigo.26
A DGAT-1 é uma enzima necessária para a síntese dos TG a partir da gordura da dieta. O pradigastat inibe esta enzima, tendo sido altamente eficaz na redução da lipémia pós-prandial. Apesar de promissor na terapêutica do SQF, o seu desenvolvimento foi suspenso pelos graves efeitos adversos gastrointestinais.10
A terapêutica genética com o alipogene tiparvovec destinada ao défice de LPL esteve disponível há alguns anos, tendo sido descontinuada pelo produtor por ausência de eficácia sustentada em conjunto com custos elevados.27 A terapêutica implicava múltiplas injecções intramusculares de um vector viral que continha uma variante da LPL com ganho de função. No entanto, após 12 semanas de tratamento, os níveis plasmáticos de TG normalizavam. A resposta imunitária contra o vector viral anulava a eficácia da terapêutica.27 Retrospetivamente, verificou-se uma redução da frequência de pancreatite aguda.14
Tendo em conta o papel da ANGPTL3 no metabolismo lipídico, esta surge como um alvo terapêutico. Actualmente, estão em fase precoce de desenvolvimento duas abordagens: o anticorpo monoclonal inibidor da ANGPTL3 evinacumab e o oligonucleótido anti-sentido para a apoC-III IONIS-ANGPTL3-LR.27
O evinacumab foi estudado em voluntários saudáveis em doses únicas crescentes até 20 mg/kg com reduções dos TG e do LDL de até 75% e 23%, respetivamente.28
O IONIS-ANGPTL3-LRx foi testado em voluntários com hipertrigliceridemia com múltiplas doses (10, 20, 40, ou 60 mg subcutâneos por semana durante 6 semanas) resultando em reduções de ≤63,1% (TG), 32,9% (LDL), 60,0% (VLDL), 36,6% (não-HDL), 25,7% (apoB-100), e 58,8% (apoC-III).29
VOLANESORSENO
O volanesorseno é um inibidor da síntese da apoC-III de segunda geração que actua nos hepatócitos. É um oligonucleótido anti-sentido que se liga selectivamente ao RNA mensageiro (mRNA) da apoC-III, o que evita a sua translação e permite a sua degradação por uma ribonuclease (Fig. 1).
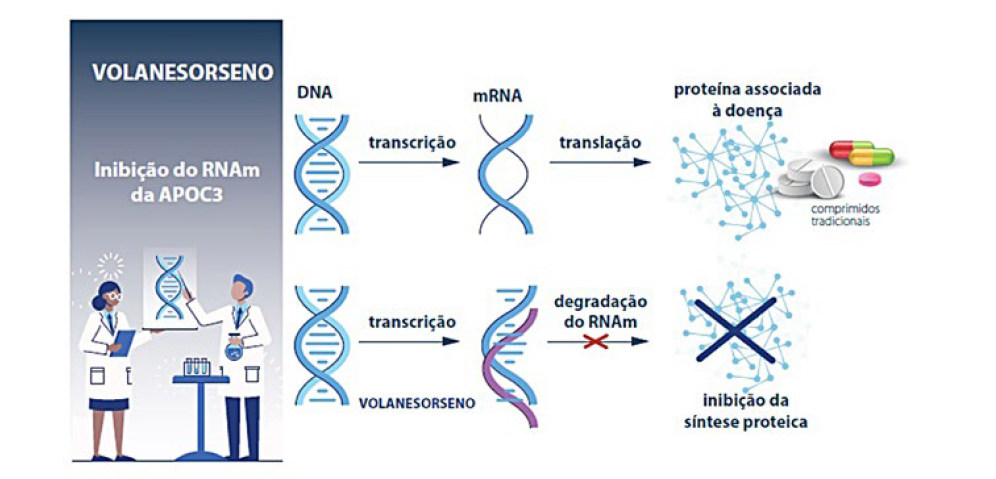
Inicialmente, colocou-se a hipótese de que a apoC-III aumentava os TG plasmáticos pela inibição da actividade da LPL, no entanto, um ensaio inicial de pequenas dimensões mostrou que o volanesorsen também reduzia os TG entre 56% e 86% em 3 doentes com SQF, o que demonstra que a apoC-III também inibe uma via de depuração de lipoproteínas ricas em TG não dependente da LPL.16
Assim, promove a clearance dos TG por vias independentes da LPL como a inibição pela apoC-III de receptores hepáticos e os receptores da proteína 1 relacionada com o receptor de LPL.30
Nos estudos de fase 1 em indivíduos saudáveis foi de-monstrado que o volanesorseno atinge o pico de concentra-ção plasmática rapidamente após a administração (2-4 horas no intervalo 50-400 mg) e os tempos de permanência curtos (6,4-8,1 horas) sugerem que a distribuição pelos tecidos é rápida. A farmacocinética é semelhante nos doentes com SQF, sendo a concentração em estado estacionário superior com doses semanais ao invés de bissemanais.
O volanesorseno liga-se fortemente às proteínas plasmáticas (>98%) independentemente da concentração com um volume de distribuição estimado nos doentes com SQF de 330L.
A semivida plasmática em voluntários saudáveis que rece-beram várias doses durou entre 11,7 e 31,2 dias, com uma eliminação sobretudo por metabolitos urinários.30
Num estudo de fase 1 em voluntários saudáveis, o volanesorseno provocou uma redução dependente da dose na apoC-III e dos TG.31Nos estudos de fase 2, demonstrou a sua eficácia a reduzir os TG em doentes com elevados níveis de VLDL com múltiplas causas. Em particular, a dose mais eleva-da de 300 mg semanais reduziu a apoC-III e os TG em 79,6% e 70,9% respetivamente.32-34
A eficácia e segurança do volanesorsen foi avaliada em dois ensaios de fase 3. No COMPASS, um ensaio randomiza-do, duplamente cego e controlado por placebo, 113 doentes com hipertrigliceridemia grave foram aleatorizados para ad-ministração de 300 mg de volanesorseno semanais versus placebo por 26 semanas. O volanesorseno mostrou uma redução nos TG de 71,8% juntamente com uma redução do risco de pancreatite aguda. Excepto pela elevada incidência de reacções cutâneas no local de administração (23,5%), o fármaco foi bem tolerado.35
No ensaio APPROACH, foram aleatorizados 66 doentes com SQF com TG em jejum ≥750 mg/dL para terapêutica com 300 mg de volanesorseno por via subcutânea semanalmente durante 52 semanas versus placebo. Foi o maior estudo até ao momento realizado nesta população. Foi obtida uma redução de TG de 77% em comparação aos valores iniciais no grupo tratado com volanesorseno em comparação com um aumento de 18% no grupo placebo. A incidência de pancreatite aguda foi também reduzida durante o tratamento (um caso no grupo de intervenção versus 4 no placebo), contudo, a baixa frequência não permite significância estatística dos resultados.
À semelhança do COMPASS, o efeito adverso mais comum foram reações cutâneas no local de administração da terapêutica (17%). A ocorrência de trombocitopénia levou à exclusão precoce de 5 doentes, que recuperaram após suspensão da terapêutica e corticoterapia.36Num estudo randomizado em 15 doentes com diabetes mellitus com hemoglobina glicada superiores a 7,5% e hipertrigliceridemia superior a 200 mg/dL, os efeitos foram acompanhados por um acréscimo na sensibilidade à insulina, com correlação significativa com a apoC-III e TG.37
A longo prazo, a eficácia do volanesorseno é sustentada no tempo. Mais ainda, o colesterol não-HDL foi reduzido em 45%, sugerindo que a redução de apoC-III conseguida com este fármaco incrementa a clearance de lipoproteínas ricas em TG nos doentes sem actividade da LPL.38
O estudo ReFOCUS realizado em doentes tratados com volanesorseno permitiu descrever uma melhoria na sua quali-dade de vida global, com um decréscimo significativo dos sin-tomas em todos os domínios estudados, incluindo sintomas físicos, emocionais e cognitivos.24
Em relação ao impacto sobre o risco cardiovascular, os dados disponíveis são limitados. Foi demonstrada uma mar-cada redução dos TG com redução moderada da Apo B com a inibição da ApoC-III enquanto a inibição da ANGP3TL resulta na redução marcada de ambos. Pode assim colocar-se a hipótese que os inibidores da ANGP3TL poderão oferecer maior redução do risco cardiovascular do que os inibidores da ApoC-III, no entanto são necessários mais estudos com estas moléculas para determinar o seu potencial nesse sentido.9
ALGORITMO DIAGNÓSTICO DA SQF
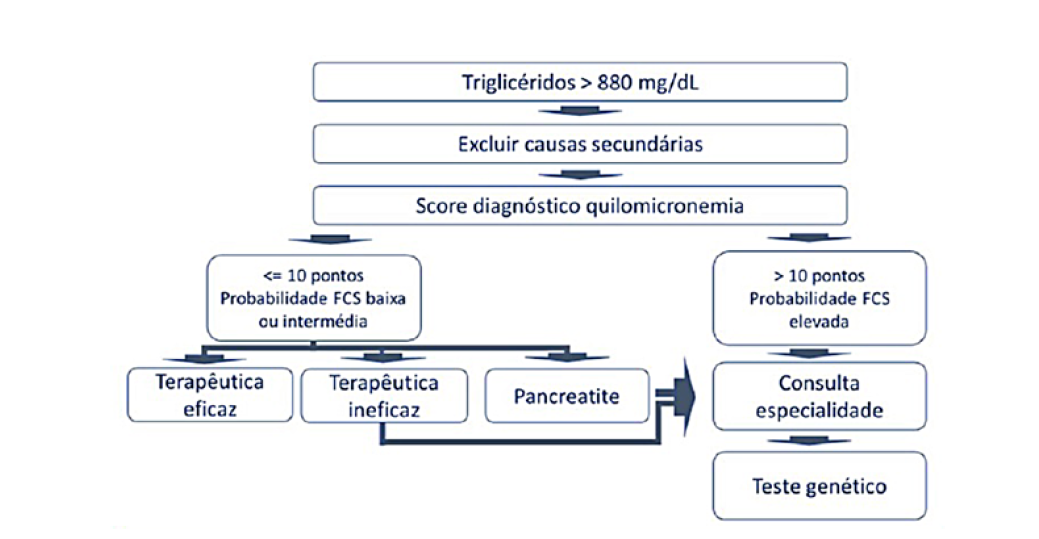
Tratando-se de uma doença rara, o diagnóstico só é possível se o clínico estiver ciente da sua existência. O conheci-mento da doença em conjunto com alguns dados da história clínica e perfil lipídico permitem validar a suspeita clínica ainda a nível dos cuidados de saúde primários. É possível assim, à luz do conhecimento atual, sugerir um algoritmo de decisão para auxiliar na marcha diagnóstica. Optamos por definir que o diagnóstico será efetuado em duas fases: a primeira em ambiente de cuidados primários, em que a suspeita clínica levanta o diagnóstico. A segunda, em ambiente hospitalar para confirmação do diagnóstico (Fig. 2).
Fase 1 - Cuidados de saúde primários
Com base no estudo de Johansen et al39 Propõe-se como limite de triglicéridos em jejum, o valor de 880 mg/dL para o diagnóstico de quilomicronemia.
Uma vez detetada a quilomicronemia, a abordagem inicial implica a exclusão de causas secundárias. Enquanto algumas, como o alcoolismo e a diabetes mellitus são rapidamente despistadas, outras doenças endócrinas ou genéticas requerem uma investigação mais aprofundada. A correção destas causas e reavaliação laboratorial favorável, pode reforçar a suspeita da etiologia secundária da hipertrigliceridemia.
O conhecimento da doença em conjunto com alguns dados da história clínica e perfil lipídico permite validar a suspeita clínica ainda a nível dos cuidados de saúde primários. Moulin et al (2018) propuseram um score diagnóstico para SQF, baseado em dados clínicos e na idade do paciente (Fig. 3). Este score foi desenvolvido e validado externamente em duas coortes italianas, obtendo-se uma sensibilidade de 88% (IC 95%: 0,97, 0,76) e especificidade de 85% (IC 95%: 0,94, 0,75). Trata-se assim de uma ferra-menta de avaliação fenotípica útil que permite selecionar os pacientes que devem ser referenciados a uma consulta de especialidade hospitalar.8

Fase 2 - consulta de Lipidologia/Especialidade
A consulta hospitalar tem como objectivos a confirmação do diagnóstico definitivo da causa de quilomicronemia (por testes bioquímicos ou genéticos), a estratificação do risco de complicações (metabólicas, cardiovasculares, pancreatite), a seleção da terapêutica apropriada e o seguimento dos doentes com SQF confirmado.
O perfil lipídico pode fazer suspeitar de SQF. Um rácio de TG/CT maior que 5 e uma ApoB menor que 100 mg/dL com TG superiores a 885 mg/dL são compatíveis com a presença de quilomicronemia e VLDL de grandes dimensões.8
Actualmente, o gold standard do diagnóstico é a identi-ficação de uma variante patogénica nos genes do complexo da LPL. A facilitação do acesso a testes de sequenciação de nova geração (NGS) virá provavelmente facilitar o diagnóstico de SQF uma vez que os genes LPL, APOC2, APOA5, GPIHBP1, LMF1 associados ao metabolismo dos quilomicra poderão ser testados com recurso a um painel de genes.9 No entanto se as mutações encontradas não tiverem sido descritas anteriormente é necessário realizar o teste da atividade da LPL para determinar se as alterações encontradas são causadoras de doença.
Conclusão
A SQF é uma doença rara cuja principal manifestação é a pancreatite aguda. O diagnóstico diferencial é dificultado pela existência de causas secundárias de hipertrigliceridemia.
Assim Moulin et al propõem que a SQF se defina como uma doença monogénica, com potencial suficiente para man-ter os níveis de TG constantemente acima de 880 mg/dL com descompensações frequentes na ausência de factores secundários óbvios. Além do défice homozigótico de LPL, consideram-se também as mutações nos genes APOC2, APOA5, GPIHBP1, LMF1 e G3PDH em homozigotia ou heterozigotia composta. Em oposição, os doentes com QMM podem ter uma combinação de variantes em heterozigotia de perda de função e/ou polimorfismos funcionais nos genes que elevam os TG o que resulta numa quilomicronemia flutuante - entre as descompensações podem ter apenas uma hipertrigliceridemia ligeira.8 O teste genético é importante para fazer a distinção entre estas duas condições.
O tratamento da SQF tem-se resumido até hoje à restrição dietética severa, que se tem revelado ineficaz na resolução do problema. Actualmente, está disponível uma terapêutica eficaz no controlo da hipertrigliceridemia e dos múltiplos sin-tomas associados, com segurança demonstrada. O diagnóstico precoce assegura o controlo metabólico e a qualidade de vida dos doentes com SQF.

