











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
A púrpura de Henoch-Schonlein (PHS) é um subtipo de vasculite cutânea de pequenos vasos que se caracteriza tipicamente pela deposição de IgA - mas também, embora menos comum, de IgM, IgG, complemente e fibrinogénio - nas paredes dos pequenos vasos da pele, glomérulos renais e trato gastrointestinal. A deposição nos vasos origina as lesões petequiais/purpúricas palpáveis, sem trombocitopenia, tipicamente nas extremidades inferiores.1-6A PHS é mais comum nas crianças (20/100 000), do que em adultos e idosos (1,3/100 000).2 Clinicamente manifesta-se pela tétrade de púrpura palpável, artrite/artralgias, dor abdominal, hemorragia gastrointestinal e lesão renal.3 O envolvimento gastroin-testinal com isquemia intestinal e perfuração são raras.10 Infeções, fármacos (sendo as principais classes conhecidas os antibióticos, anti-TNF e vacinas) e neoplasias são alguns dos triggers reconhecidos, embora a fisiopatologia ainda não esteja totalmente esclarecida.3,9A ordem de apresentação varia, sendo que geralmente se manifesta inicialmente com petéquias/púrpura e queixas articulares, o envolvimento intestinal surge uma semana após, e o atingimento renal é encontrado entre 2 a 6 meses desde o início das lesões cutâneas.10 O atingimento cutâneo é geralmente bilateral e simétrico, sendo os membros inferiores e a região nadegueira as zonas preferenciais, mas não limitado a estas podendo atingir todo o corpo.4 O diagnóstico é clínico sendo que os critérios de diagnóstico vigentes foram publicados em 2010 (EULAR/PRINTO/PRES), apresentando uma sensibilidade de 99% e especificidade de 86%.3,8A biópsia cutânea é gold-standard todavia insuficiente, isoladamente, para o diagnóstico dado que outras patologias se podem manifestar com depósitos vasculares de IgA (outras síndromes vasculíticos, estase venosa). Alguns autores defendem inclusivamente que a biópsia cutânea deva ser reservada para os casos em que o diagnóstico não seja possível pelos achados clínicos ou se a apresentação é atípica ou incompleta.3 Alguns dos diagnósticos diferenciais a contemplar são as vasculites de hipersensibilidade, outras vasculites de pequenos vasos (granulomatose com poliangiite, granulomatose eosinofílica com poliangiite, crioglobulinemia ou vasculites secundárias a conectivites) e doenças infecciosas (hepatite B ou C).1 O tratamento é geralmente sintomático quando não existe atingimento renal. Os corticoides proporcionam uma rápida resolução no caso de atingimento articular, dor abdominal e atingimento renal. Todavia, neste último caso, outros imunossupressores como micofenolato de mofetilo ou a ciclosporina A são as escolhas mais adequadas.3 Geralmente a PHS tem uma evolução benigna e autolimitada. A morbilidade relaciona-se com evolução para a doença renal crónica, sendo que em 7% dos casos há desenvolvimento de síndrome nefrítica ou nefrótica sendo que em 1% dos casos há evolução para doença renal terminal.3 Em um terço dos casos há recorrência sendo também mais frequente em doentes com atingimento renal.1 É importante contemplarmos este diagnóstico na população adulta pois a morbimortalidade associada ao envolvimento renal é mais frequente, ao contrário do que acontece na população pediátrica. Também nos adultos, a associação com neoplasia é mais frequente, com uma incidência de 2%-5%, principalmente neoplasias hematológicas.3 e por isso esta deve ser excluída.
Caso Clínico 1
Apresentamos um homem de 64 anos, autónomo nas ati-vidades de vida diária. Recorreu ao Serviço de Urgência do Centro Hospitalar e Universitário do Porto por rash petequial com 11 dias de evolução desde as regiões acrais e topografia ascendente e coalescência das lesões; concomitantemente com diarreia sem sangue ou muco. Sem artralgias ou hematúria. Sem infeções víricas prévias. Sem introdução de novos fármacos. Doente com antecedentes de relevo de hipertensão arterial e dislipidemias controladas com amlodipina em associação com indapamida e rosuvastatina; medicado com valproato de sódio, escitalopram e memantina por perturbação do humor e síndrome demencial incipiente; hiperplasia benigna da próstata medicado com tansulosina. Ao exame objetivo, apresentava petéquias palpáveis nos membros, extremidades e tronco, sem outras alterações. Analiticamente sem qualquer alteração à exceção de elevação ligeira da PCR (27,8 mg/L, valores de referência (vr) 0-5 mg/L) e ferritina (316 mcg/dL, vr 75-150 mcc/dL). Estudo imunológico também sem alterações. Serologias víricas negativas para VIH, VHB, VHC, EBV e CMV. Realizou ecografia abdominal com sinais de colite, nomeadamente espessamento de segmentos da parede intestinal e hiperecogenecidade da gordura adjacente. Assumiu-se diagnóstico provável de púrpura de Henoch-Schonlein e teve alta com prednisolona 60 mg durante 3 dias, com desmame progressivo. O resultado da biópsia cutânea, viria a confirmar o diagnóstico, revelando a presença de vas-culite leucocitoclástica com depósitos de IgA e fibrinogénio na parede dos pequenos vasos (Fig.s 1 a 3). O doente evoluiu com episódio inaugural de diarreia sanguinolenta e dor abdominal ligeira ao terceiro dia de prednisolona, motivo pelo qual foi reavaliado. Analiticamente não havia alterações de relevo. Dada a estabilidade às 72 horas, teve alta, optando-se por um esquema de corticoterapia mais longo e desmame mais lento, iniciando-se 40 mg prednisolona oral. Recorreu novamente ao SU após 2 dias por novo episódio abundante de diarreia sanguinolenta com dor abdominal associada, mesmo sob corticoterapia. Analiticamente apresentava aumento dos parâmetros inflamatórios (PCR 84 mg/L, vr 0-5 mg/L) e lesão renal aguda (creatinina e ureia séricas 1,29/89 mg/dL) e sedimento urinário com proteinúria de 1,59 g/24 horas e vestígios de sangue, motivo pelo qual é admitido em internamento hospitalar. Realizou angiografia por tomografia computorizada (angio-TC) abdomino-pélvico que revelou espessamento de ansas de delgado com várias imagens de depleção em ramos segmentares das artérias mesentéricas compatíveis com a hipótese de envolvimento intestinal por vasculite de pequenos vasos. Foi tratado com metilprednisolona 1 g EV durante 3 dias e, dado o atingimento renal, iniciou 1 g de ciclofosfamida EV e MESNA. Evoluiu com boa resposta global, desaparecimento de dor abdominal, ausência de expressão sistémica relevante da inflamação e com tolerância da dieta oral. Apresentou melhoria paulatina da função renal até normalização e redução da proteinúria para 1 g/24h. Lesões cutâneas mantiveram aspeto, sem agravamento. Observada melhoria imagiológica em TC abdominal mostrando resolução completa dos achados de isquemia que envolviam o intestino delgado e cólon descendente. Cumpriu 7 dias de predniso-lona 1 mg/kg/dia (80 mg/dia) tendo alta com 0,5 mg/kg/dia (40 mg/dia). Evolução favorável na reavaliação em ambulatório, tendo cumprido 3 ciclos de ciclofosfamida e 9 meses de corticoterapia. Após 2 anos de acompanhamento, mantém-se sem recidiva e sem evolução para doença renal crónica, com creatinina e ureia séricas normais e mantendo níveis de microalbuminúria <300 mg/24h.
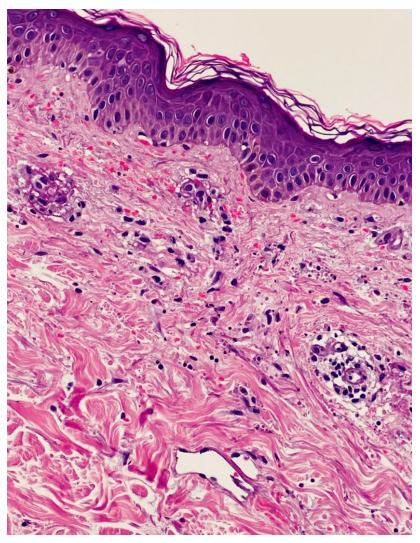
Figura 1: HE 20 x - Pele com vasos do plexo superficial com necrose fibrinóide da parede, ligeiro infiltrado inflamatório com neutrófilos, abundante leucocitoclasia e extravasamento eritrocitário.
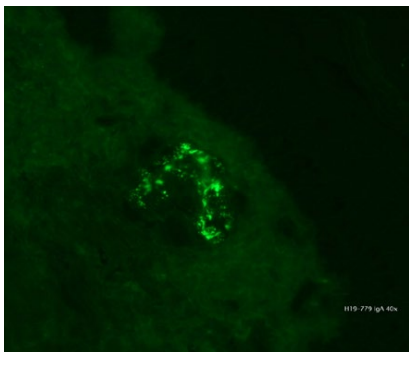
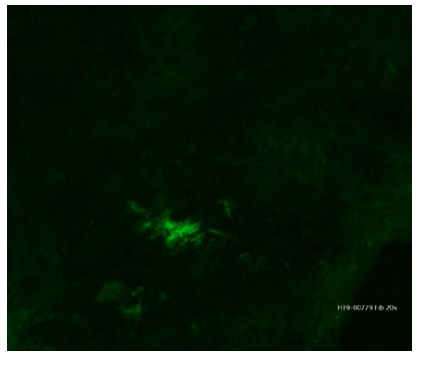
Caso Clínico 2
Apresentamos uma mulher de 90 anos, autónoma para as atividades de vida diária. Recorreu ao Serviço de Urgência por lesões petéquias e púrpura maculopapular, com 2 dias de evolução nos membros inferiores e antebraços. Sem febre, queixas abdominais ou artralgias. Sem história de infeção recente ou fármacos de novo. Doente com antecedentes de dislipidemia, insuficiência venosa crónica, doença de Meniére e aneurisma da aorta abdominal já intervencionado, medicada com atorvastatina, bioflavonóides, betahistina e ácido acetilsalicílico. Analiticamente, não tinha alterações salvo VS elevada 109 mm/h (vr 0-5 mm/h) e PCR 17,96 mg/L (vr 0-5 mg/L). Realizou biópsia cutânea e teve alta para o domicílio. Na reavaliação em ambulatório um mês após, dado a progressão das lesões cutâneas e edemas periféricos de novo, analiticamente, com aumento da creatina e ureia séricas (1,46/60 mg/dL) em relação ao seu basal (creatinina de 0,8 mg/dL), com proteinúria subnefrótica associada, foi admitida no internamento. O resultado da biópsia cutânea que ficou em curso descreveu vasculite leucocitoclástica embora sem depósitos de imunoglobulinas ou complemento na imunofluorescência, mas com a presença de anticorpo anti-fibrinogénio na parede de alguns vasos. Evoluiu com hematúria e dismorfia eritrocitária e agravamento da proteinúria para níveis nefróticos. Do estudo complementar no internamento, apresentava aumento de IgA e IgG e estudo imunológico com ANA, ANCA, crioglobulinas e anti-membrana basal do glomérulo negativos. Serologia VIH, HBV e HCV negativos. TC tóraco-abdomino-pélvico sem lesões sugestivas de neoplasia e citometria de sangue periférico excluiu doença linfoproliferativa. Sem semiologia que motivasse a realização de outros exames complementares, assumiu-se o diagnóstico de púrpura de Henoch-Schonlein e pela disfunção renal iniciou corticóide em alta dose (1 mg/kg/dia). Por hipertensão arterial enquadrada no atingimento renal, iniciou ARA II. Evoluiu favoravelmente com resolução das lesões e melhoria da função renal, embora mantendo proteinúria em nível sub nefrótico (2,4 g/dL). Na reavaliação em ambulatório, manteve prednisolona em desmame lento, durante um ano, sem reci-diva, mantendo o valor de creatinina sérica basal em 1,3 mg/dL, mas com diminuição da proteinúria, para microalbuminúria inferior a 100 mg/dia.
Discussão
Em nenhum dos casos apresentados foram identificados precipitantes, nomeadamente infeções do trato respiratório superior. Apesar de nos dois casos existirem fármacos descritos como possíveis indutores de vasculite de IgA (valproato de sódio e ácido acetilsalicílico, respetivamente), esta etiologia foi afastada, não só pela ausência de temporalidade entre a clínica e a toma dos fármacos, mas sobretudo pela evolução favorável dos dois doentes com o tratamento instituído, sem recidiva após o desmame do corticóide, mesmo com a manutenção dos fármacos potenciais.8,9 Os dois casos evoluíram com disfunção renal e proteinúria em níveis sub-nefrótico e nefrótico. No primeiro caso, ocorreu ainda isquemia intestinal, confirmado por angio-TC, uma apresentação mais rara e severa que se manifestou com dor abdominal violenta e com diarreia sanguinolenta.10 Já no segundo caso, a clínica era mais parca, porém, e apesar da biópsia cutânea não ser típica (cujo resultado poderá ter sido afetado pelo timing da sua realização, que idealmente deverá ser em lesões com menos de 24 horas de evolução),2 o estudo imunológico e as serologias víricas foram negativos, assim como também se excluiu a etiologia neoplásica (que acontece em 3% a 85% das vasculites). No primeiro caso houve resolução da lesão renal, sendo que no segundo, a doente manteve algum grau de lesão renal, apresentando durante o internamento fatores de mau prognóstico - síndrome nefrótica e hipertensão arterial.3,8
Conclusão
A PHS é uma entidade clínica bem reconhecida e docu-mentada na população pediátrica mas muito menos comum em adultos, em especial nos idosos. É geralmente auto-limitada ao contrário de outras vasculites sistémicas. O diagnóstico é clínico sendo que os achados na biópsia cutânea são específicos, mas não sensíveis, admitindo especial importância no caso de apresentações atípicas ou incompletas. A morbilidade a curto prazo tende a relacionar-se com o atingimento gastrointestinal e a longo prazo com o envolvimento renal. Em um terço dos doentes há recorrência. Esta entidade deve ser suspeitada na população adulta e idosa, pois, o seu diagnóstico precoce poderá prevenir morbimortalidade, pelo atingimento renal, a longo prazo.
Declaração de Contribuição / Contributorship Statement:
S. M. Azevedo, S. I. Rocha - Análise e Recolha de Dados, Escrita e Interpretação , Revisão crítica.
M. V. Bertão, A. Ferreira - Análise e Recolha de Dados, Revisão crítica.

