











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
A doença de Gaucher (DG) é uma doença genética rara, com uma incidência estimada em 1 :40 000 nados vivos, sendo a doença mais frequente do grupo das Doenças Lisossómicas de Sobrecarga (DLS-**92921 A incidência é consideravelmente maior na população judaica Ashkenazi (1 :800 nados vivos).1 Recebeu o nome de doença de Gaucher devido ao estudante de medicina francês que a descreveu pela primeira vez em 1882: Philippe Gaucher.2•3Em Portugal, existem cerca de 140 casos diagnosticados, no entanto, tendo em conta a dificuldade no seu diagnóstico, acreditase que muitos estejam ainda por identificar.4 Tem padrão de hereditariedade autossómico recessivo e caracteriza-se por mutações no gene GBA e consequente défice ou diminuição da actividade da enzima glucocerebrosidase (também conhecida como -glucosidase ácida).3·5Mais de 200 mutações foram encontradas no gene codificador da glucocerebrosidase lisossómica; contudo, reveste especial importância a presença do alelo N370S por estar associado à doença não-neuropática (DG tipo 1) e do alelo L444P que está relacionado com sintomas neurológicos precoces (DG tipo 2).3·6
Como resultado da mutação, há acumulação de glucocerebrosídeos nos macrófagos (células de Gaucher), que se depositam em múltiplos órgãos (principalmente baço, fígado, medula óssea, cérebro e osteoclastos).3 É uma doença grave, crónica, progressiva e multissistémica que pode apresentar um conjunto muito variado de sinais e sintomas comuns a outras doenças.5·6
Clinicamente, é classificada em 3 subgrupos.4-6A tipo 1, conhecida como forma adulta ou não-neuropática, é a mais comum, representando >90% de todos os casos de DG.
Tem uma sobrevida próxima à população geral e caracteriza-se por pancitopenia, hepatoesplenomegalia, hipertensão pulmonar e patologia óssea (osteoporose, osteoartrose, osteonecrose, fracturas patológicas, deformidade em frasco Erlenmeyer, dor óssea e/ou atraso de crescimento). A tipo 2, conhecida como forma infantil ou neuropática aguda, tem uma sobrevida inferior a 3 anos de idade. Neste subtipo predominam os sintomas neurológicos graves como apraxia oculomotora, convulsões, hipertonia, mioclonias e atraso mental severo, com apneias e insuficiência respiratória.
A tipo 3, apelidada de forma juvenil ou neuropática crónica tem uma sobrevida que ronda os 40 anos, com sintomas neurológicos mais frustes e tardios (parkinsonismo e perturbações no movimento ocular).5·6
O diagnóstico confirma-se pelo doseamento enzimático da actividade da glucocerebrosidase nos leucócitos de sangue periférico ou fibroblastos. Adicionalmente, é essencial uma colheita de história pessoal e familiar pormenorizada, a realização de um exame físico rigoroso e a solicitação de análises e exames imagiológicos, para avaliar, por exemplo, a presença de citopenias, organomegalias e atingimento ósseo.3 O tratamento da DG tipo 1 tem dois modos: terapêutica de substituição enzimática (TSE) ou terapêutica reductora de substrato (TRS).4•5Na TSE com imiglucerase, velaglucerase ou taliglucerase, fornecemos formas recombinantes da enzima glucocerebrosidase que degradam os glucocerebrosídeos, reduzindo a sua acumulação nas células e órgãos. A administração destes fármacos é endovenosa quinzenal a mensal. Na TRS, com miglustato e eliglustato (administração oral e diária), é inibida a glucosilceramida sintetase (enzima da reacção oposta), impedindo a produção de mais glucocerebrosídeos. Embora também se possa utilizar a reposição enzimática na DG tipo 2 e tipo 3, os resultados não são tão encorajadores e o prognóstico continua a ser muito sombrio.3
Com este estudo, pretende-se caracterizar a população de doentes com DG seguida em consulta e entender o impacto do tratamento na progressão da doença.
Material e Métodos
Desenho do Estudo:
Realizou-se uma série de casos de doentes com doença de Gaucher em terapêutica de substituição enzimática, seguidos em consulta de Medicina Interna no Hospital de Braga em 2019. O estudo foi retrospectivo e longitudinal, uma vez que foram colhidos dados dos doentes desde a data do diagnóstico da doença até 2019. Obteve-se uma amostra final de 5 doentes.
Recolha de Informação:
Procedeu-se ao levantamento e análise dos cinco processos clínicos sob a forma de suporte papel e electrónico. Todos os doentes forneceram consentimento informado para a utilização e divulgação dos seus dados pessoais e clínicos. O projecto foi avaliado pelo encarregado de protecção de dados e aprovado pela Comissão de Ética do Hospital de Braga. Os dados foram registados num formulário específico que garantia a confidencialidade dos doentes. Foram analisados os seguintes dados: género, idade, idade ao diagnóstico, idade ao início da TSE, sintomas pré-tratamento, método de diagnóstico da doença, subtipo de DG, história familiar, tipo de TSE, duração de tratamento, valores de hemoglobina e plaquetas antes e durante TSE, dimensões do fígado e baço antes e durante TSE, valores dos biomarcadores (quitotriosidase e ácido resistente ao tártaro - TRAP) antes e durante TSE.
Análise dos Dados:
A análise univariada foi utilizada para a análise descritiva dos dados: média (M), máximo, mínimo e mediana (Mdn). A análise estatística foi realizada com recurso ao Statistica/ Package for the Social Sciences®(SPSS) versão 20.0.
Resultados
Obteve-se uma amostra de cinco doentes com DG, seguidos em consulta de Medicina Interna, sob TSE (Tabela. 1 ). Todos eram do sexo feminino e apresentavam DG do tipo 1. A idade ao diagnóstico variou entre 6 e 35 anos (média=23 anos, mediana 16 anos).
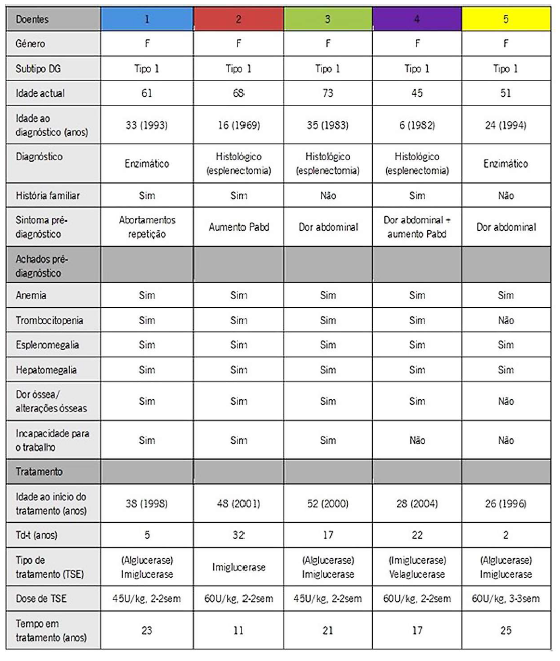
Relativamente à sintomatologia antes do diagnóstico, três doentes apresentavam dor abdominal, dois aumento do volume abdominal e um abortamentos espontâneos de repetição. Nesta última doente (doente 1), demorou-se cerca de 10 meses para efectuar o diagnóstico. Nas restantes doentes, não foi possível apurar com precisão o tempo decorrido entre o início dos sintomas e o diagnóstico, uma vez que os registos em formato papel referem organomegalias ao exame físico desde idade muito precoce (primeiros anos de vida). Em três doentes o diagnóstico foi histológico em peça de esplenectomia (nas décadas 60-80), sendo que dois tiveram diagnóstico enzimático (década de 90). Três das 5 doentes apresentam história familiar de DG. De salientar que, previamente à TSE, todas apresentavam anemia e hepatoesplenomegalia. Quatro das 5 doentes tinham trombocitopenia e a mesma proporção tinha dor/patologia óssea.
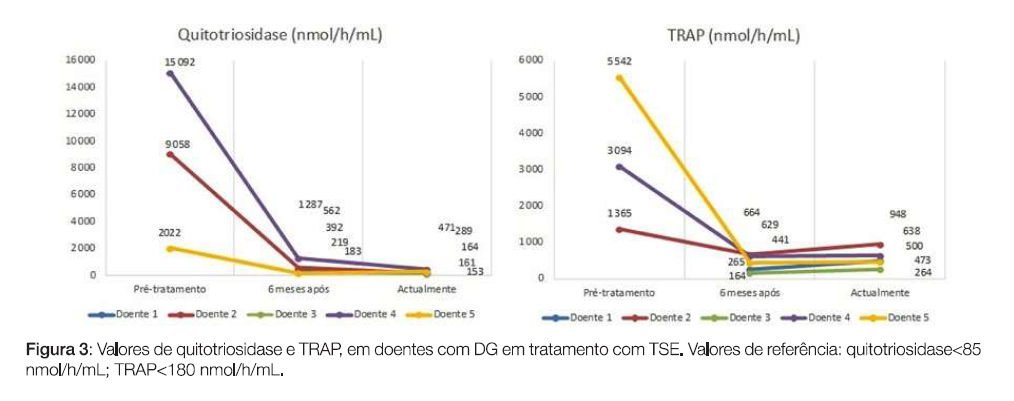
Relativamente ao tratamento, quatro doentes estavam medicadas com imiglucerase, três das quais haviam iniciado TSE com alglucerase. Uma doente estava actualmente medicada com velaglucerase (iniciou TSE com imiglucerase, emigrou e no regresso foi tratada com velaglucerase). A idade de início de TSE variou entre os 26 e 52 anos (média=38 anos, mediana=38 anos). Após 6 meses de tratamento, verificou-se melhoria da anemia e trombocitopenia para valores próximos dos normais (Fig. 1) e redução do volume do baço e fígado (Fig. 2). De ressalvar que na Fig. 1 apenas constam as doentes que apresentavam trombocitopenia previamente à TSE, bem como na Fig. 2 não constam as doentes cujo diagnóstico foi efectuado por histologia (após esplenectomia). Em média, o valor de hemoglobina subiu 1 ,9 g/dl em 6 meses de tratamento e a contagem de plaquetas subiu em média 75 000 plaq/ul. Houve uma diminuição de 1,7 cm de maior eixo longitudinal do fígado em 6 meses de tratamento e de 6,5 cm do baço. Verificou-se ainda redução dos níveis de alguns marcadores biológicos, como p-D-quitotriosidase e fosfatase ácida resistente ao tártaro (TRAP) (Fig. 3). Quatro das doentes tinham alterações na densitometria óssea compatíveis com osteopenia/osteoporose previamente ao tratamento, sendo que actualmente, sob TSE, todas apresentam valores de densidade mineral óssea normal em relação ao esperado para uma população da mesma idade e sexo (z-score>-0,5).
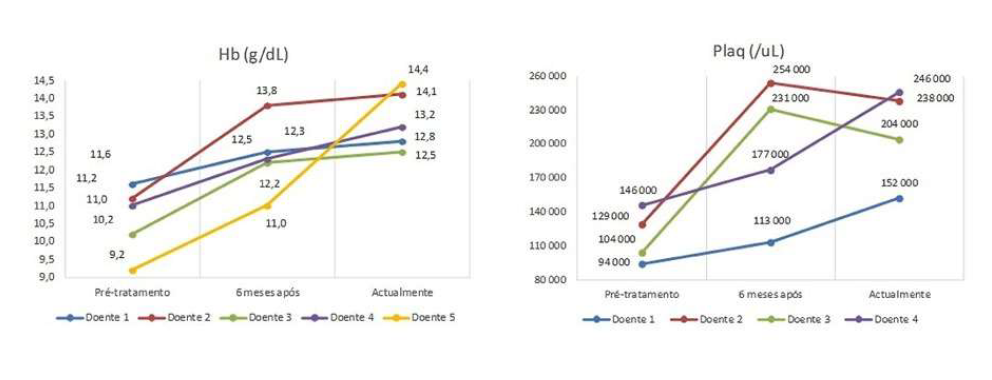
Figura 1: Valores de hemoglobina e plaquetas ao longo do tempo, em doentes com DG em tratamento com TSE.
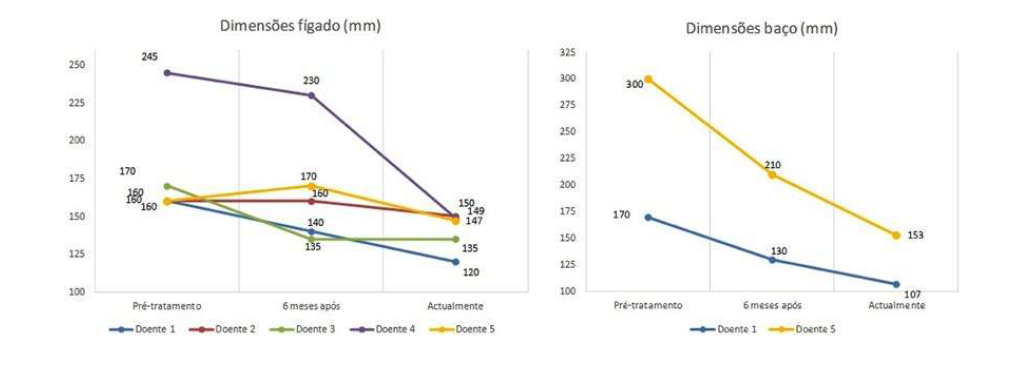
Figura 2: Dimensões (maior eixo longitudinal) do fígado e baço ao longo do tempo, em doentes com DG em tratamento com TSE.
Discussão
A doença de Gaucher é uma doença genética, que se pode apresentar em qualquer idade. No entanto, uma parte significativa dos casos só é diagnosticada na idade adulta.
De acordo com dados recentes, sabe-se que uma em cada seis pessoas espera sete anos pelo diagnóstico.4 Antigamente, o diagnóstico era muitas vezes histológico, após esplenectomia ou por biópsia da medula óssea, devido à ausência de alguns exames complementares de diagnóstico. Actualmente, com o avançar da ciência e tecnologia, o diagnóstico é fácil, rápido e praticamente indolor, obtendo-se através duma simples gota de sangue, retirada por uma picada no dedo.3 O essencial é a suspeita clínica e conhecimento da DG, para realizar o diagnóstico atempado. Atendendo a que se trata de uma doença de transmissão genética, devem ser realizados rastreias e aconselhamento genético aos familiares directos.4 Os doentes de Gaucher são habitualmente seguidos por pediatras, internistas ou hematologistas, atendendo às manifestações e ao carácter multissistémico da doença, mas qualquer médico poderá fazer o diagnóstico. Em concordância com os resultados deste estudo, a DG tipo 1 é o subtipo mais frequente e as manifestações clínicas típicas são hepatoesplenomegalia, citopenias e patologia óssea.3·5,6Verificou-se que doentes esplenectomizados desenvolviam doença óssea mais precocemente comparativamente aos doentes com DG não esplenectomizados.1 Nas doentes em estudo, as 3 doentes esplenectomizados desenvolveram deformidades em frasco de Erlenmeyer mais precocemente que as restantes. Uma delas desenvolveu osteonecrose da cabeça do fémur, com necessidade de intervenção cirúrgica precoce, motivo pelo qual iniciou tratamento com Velaglucerase após discussão com peritos internacionais. De salientar que durante o tratamento, houve uma melhoria da densidade mineral óssea das doentes. Todas as doentes seguidas em consulta iniciaram TSE em idade adulta. Sendo a DG clinicamente heterogénea e classificada em três tipos, o seu prognóstico pode variar de benigno a extremamente grave. De qualquer forma, pela sua cronicidade e progressão, se não for tratada, pode levar a uma sintomatologia exuberante, incapacitante e a morte precoce. Quando o tratamento é atempadamente instituído, os resultados são habitualmente favoráveis, com recuperação dos valores hematológicos, redução do volume das organomegalias e prevenção das lesões ósseas, melhorando consideravelmente a qualidade de vida dos doentes.7 Uma vez que as células de Gaucher produzem quitotriosidase, a sua monitorização foi utilizada para avaliar a eficácia do tratamento e o seu valor prognóstico.8,9No nosso estudo, em 6 meses de TSE, verificou-se em média uma redução de 89% do valor de quitotriosidase. A fosfatase ácida resistente ao tártaro (TRAP}, por sua vez, é um marcador indirecto e não específico de armazenamento lipídico, pelo que valores elevados podem reflectir excesso de lípidos na DG.8,10,11Tal como seria de esperar, os valores de TRAP também reduziram com o TSE. O doseamento da glucosil-esfingosina (Lyso-GB1) é um método desenvolvido recentemente e considerado actualmente como o mais preciso na monitorização da resposta ao tratamento12 ; no entanto, ainda não existem dados em Portugal que permitam uma análise longitudinal. Neste estudo, foi possível corroborar os benefícios da TSE na melhoria das citopenias (anemia e trombocitopenia), na redução do tamanho do fígado e baço e no melhor controlo da doença, com descida dos níveis dos marcadores quitotriosidase e TRAP. As doentes com incapacidade para o trabalho, após início da TSE, tornaram-se activas e melhoraram a sua qualidade de vida.
Conclusão
A DG é uma doença grave, crónica e multissistémica que se pode apresentar com um conjunto variado de sinais e sintomas que são comuns a outras doenças. Além de rara, apresenta-se de forma gradual, o que explica muitas vezes o atraso no seu diagnóstico.
O tratamento permite franca melhoria da qualidade de vida dos doentes e do prognóstico da doença, pelo que é fundamental promover o conhecimento para esta doença e actuar de forma célere no seu diagnóstico e tratamento.

