











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
A relação entre disfunção hematológica e síndromes autoinflamatórias sistémicas encontra-se atualmente bem documentada, ficando por esclarecer a relação fisiopatológica entre as duas entidades.1 Um exemplo descrito desta associação é a ocorrência de síndrome mielodisplásica (SMD) em idosos com policondrite recidivante (PR), como evidenciado num estudo retrospetivo de 20162 que analisou 142 doentes com PR, tendo sido identificado um subgrupo com pior prognóstico: homens, idade superior a 55 anos e SMD associado. A relação entre estas entidades encontra-se agora explicada pela descoberta da síndrome VEXAS em
2020 por Beck et al,1 ao identificar 25 homens com mutações somáticas que envolvem o codão metionina-41 (p. Met41) do UBA1 - que codifica a maior enzima iniciadora da ubiquitinação. O nome atribuído a esta doença, o acrónimo VEXAS, pretende incluir as principais características inicialmente identificadas: presença de vacúolos nas células precursoras mieloides e eritroides de aspirados da medula óssea (V), enzima ativadora E1 necessária ao início da ubiquitinação intracelular (E1), ligado ao cromossoma X (X), presença de autoinflamação (A) e de mutação somática (S). A descoberta da VEXAS é também notória pelo uso de uma abordagem orientada por genótipo, que permitiu uma maior compreensão do mecanismo fisiopatológico subjacente e avanço científico para o seu tratamento.1,3
GENE UBA1 E FISIOPATOLOGIA
O principal mecanismo fisiopatológico subjacente à sín-drome VEXAS é a ocorrência de mutações somáticas do gene UBA1 nos precursores das células hematopoiéticas. Localizado no cromossoma X, o gene UBA1 codifica a enzima E1, responsável pela ativação da ubiquitina - o primeiro passo na ubiquitinação das proteínas a nível intracelular.1,3,4A ubiquitinação consiste num tipo de modificação póstranslacional das proteínas, necessária para regular diversos aspetos da homeostasia celular como vias de sinalização ou a degradação das proteínas através do sistema proteossoma e lisossoma.
Existem duas formas de expressão do gene UBA1, a isoforma nuclear (UBA1a) e a isoforma citoplasmática (UBA1b), encontrando-se esta última associada a VEXAS quando a sua função se encontra comprometida.1,3
Inicialmente foram descritas três mutações deste gene que envolvem o codão metionina-41 (p. Met41). Estas ocorrem por substituições de nucleótidos que originam uma isoforma alternativa (UBA1c) com atividade catalítica reduzida.1,3A diminuição da expressão de UBA1b leva à acumulação das proteínas por erro na ubiquitinação e à ativação do sistema imune inato, resultando num aumento da produção de citocinas inflamatórias (por exemplo: interleucina-6 (IL-6), interleucina-8 (IL-8), interferão gama e fator de necrose tumoral (TNF)), que está na base de um estado autoinflamatório sistémico.1,3,5
Atualmente já se conhecem novas mutações associadas à VEXAS, a maioria envolvendo substituições da metionina-41 (p. Met41) e todas associadas a uma perda de função de UBA1b.4 Hoje, sabe-se que a mutação UBA1b (p. Met41Val) foi a que se traduziu em clínica mais grave e pior prognóstico.6
No primeiro estudo elaborado por Beck et al,1 as mutações foram encontradas em mais de metade das células precursoras hematopoiéticas, mas estavam restritas à linhagem mieloide no sangue periférico, não se encontrando presentes nos linfócitos ou fibroblastos. São necessários estudos adicionais para compreender o mecanismo deste processo.
EPIDEMIOLOGIA
A prevalência da síndrome VEXAS não é totalmente conhecida, tendo em conta a sua identificação recente e a constante atualização dos critérios de diagnóstico. Num estudo realizado em 2022 numa população superior a 160 000 pessoas,7 Beck et al estudaram a prevalência de variantes somáticas do UBA1 associadas a manifestações sugestivas da doença, estimando que esta atingirá aproximadamente 1 em 14 000 indivíduos, elevando-se para 1 em 4249, em homens com idade superior a 50 anos (semelhante, por exemplo, à hemofilia A no sexo masculino).
Sendo uma doença ligada ao cromossoma X, foi inicialmente descrita como exclusiva do sexo masculino ou apenas presente em mulheres com aneuploidia do cromossoma X.1,7De facto, trata-se de uma patologia praticamente exclusiva dos homens, contudo, com o aumento de estudos e divulgação científica disponível, já foram descritos casos em mulheres sem aneuploidias.8,9A doença surge habitualmente entre a 5ª e a 7ª década de vida, com uma idade média na altura do diagnóstico entre os 64 e 74 anos.1,4,10
Em Portugal, a prevalência é igualmente desconhecida, tendo em conta o número reduzido de publicações científicas relacionadas com esta patologia. Uma carta ao editor publicada na Ata Médica Portuguesa em maio de 2023, divulga uma série de 7 indivíduos com síndrome VEXAS seguidos no Centro Hospitalar Universitário de Santo António, analisando as suas principais características clínicas e laboratoriais.11
COMO RECONHECER E DIAGNOSTICAR VEXAS?
VEXAS é uma doença sistémica que se caracteriza por inflamação grave e progressiva e que apresenta um grande espetro de manifestações reumatológicas e hematológicas.12Por este motivo, é frequente a descrição de patologias concomitantes nos doentes com VEXAS, nomeadamente síndrome de Sweet, PR, doença de Still, poliarterite nodosa, arterite de células gigantes,1 bem como psoríase, polimialgia reumática, sarcoidose7e síndrome de ativação macrofágica.13
MANIFESTAÇÕES CLÍNICAS
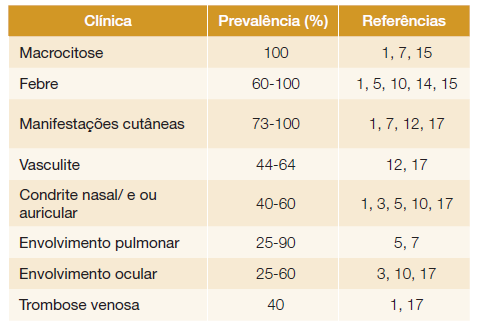
A febre é o principal sintoma, descrito em 60%-100% dos casos1,5,10,14,15a par das manifestações cutâneas presentes em 73%-100% dos casos, de acordo com vários estudos realizados.1,7,12,16,17As lesões cutâneas são tipicamente nodulares, eritematosas, dolorosas e não pruriginosas, localizando-se sobretudo no tronco e extremidades.18Também estão descritas pápulas, máculas, púrpura ou livedo.14 Uma pista diagnóstica é a reação cutânea no local de injeção em doentes tratados com antagonistas do recetor da interleucina-1 (Anakinra).18Histologicamente, a maioria apresenta dermatose neutrofílica associada a infiltração clonal mieloide e vasculite leucocitoclástica.16,19
O envolvimento ocular ocorre em 25%-60%3,10,17e as principais manifestações incluem inflamação idiopática da órbita, inflamação periorbital, uveíte, episclerite, blefarite e ainda vasculite da retina.1,10,12,16
Para além da febre, podem estar presentes perda ponderal, cansaço, mialgias e suores noturnos.5,15,16As linfadenopatias estão descritas em 60% dos casos e com múltiplas localizações.16
Quanto às manifestações reumatológicas, estas englobam artralgias, condrite, vasculite ou miosite. A condrite nasal e/ou auricular está presente em 40%-60% dos doentes, sendo que, no relatório inicial de Beck et al, cerca de 60% dos doentes VEXAS cumpriam critérios de diagnóstico para PR.1 De facto, a PR associada a VEXAS distingue-se da PR clássica porque à primeira associam-se outras manifestações como infiltrados pulmonares e alterações hematológicas, e é raro afetar a traqueia e grandes vias aéreas.3,6,10Para além das artralgias e mialgias já referidas, podem também existir como manifestações de VEXAS rigidez articular e artrite generalizada em cerca de 60% dos doentes.7,10,17
Estão descritos fenómenos vasculíticos em 44%-64% dos doentes com VEXAS.12,17A vasculite de pequenos vasos é a mais comum, com envolvimento mais frequente a nível cutâneo ou brônquico, mas também renal. Também já estão descritos casos de vasculite de médios e grandes vasos,12,17bem como casos de arterite de células gigantes, poliarterite nodosa,1 vasculite mediada por IgA, ou vasculite associada a ANCA.19
O envolvimento pulmonar pode ocorrer, estando descritas alterações imagiológicas em cerca de 25%-90% dos casos.5,7Contudo, os sintomas respiratórios não são comuns, ocorrendo em menos de metade dos doentes, sendo os mais referidos a dispneia e a tosse. As alterações podem variar ao longo do tempo e muitas vezes mimetizam infeção respiratória e insuficiência cardíaca.20Estão ainda descritos na literatura casos de pneumonia intersticial idiopática associada a VEXAS.14
Quanto às manifestações hematológicas, sabe-se que um dos principais marcadores da doença é a macrocitose, que está descrita como o primeiro sinal de disfunção hematológica e surge habitualmente após o início dos sintomas.7 Numa série de 16 doentes descrita por Obiorah et al,15 a anemia macrocítica estava presente em 100% dos doentes, 56% já dependentes de transfusão no momento do diagnóstico, e a trombocitopenia em 50%. Neste estudo foi identificado um padrão de disfunção medular progressiva de acordo com a evolução das citopenias. A primeira fase caracteriza-se pela presença de anemia macrocítica moderada sem outras citopenias, sendo que passados cerca de dois anos, a anemia macrocítica agrava-se e surge trombocitopenia ligeira. Cinco anos após a primeira alteração, o doente fica dependente de transfusões e a contagem de plaquetas diminui progressivamente.
As citopenias e a evolução para SMD é comum nos doentes com VEXAS. No estudo mencionado,15 38% dos doentes apresentavam SMD refratário à terapêutica, contudo sem progressão para SMD de alto risco ou leucemia mieloide aguda (LMA). De facto, quando presente, trata-se de SMD de baixo risco, com IPSS-R baixo, de acordo com os critérios da Organização Mundial de Saúde (OMS) propostos em 2016, com cariótipo normal e baixas contagens de blastos.15,21,22Estão ainda descritos casos de mieloma múltiplo,12,15,23gamapatia monoclonal de significado indeterminado7,15,24e leucemia linfocítica crónica15 nos doentes com VEXAS. Mais raramente pode ocorrer leucemia mielomonocítica crónica.7,25
Os eventos trombóticos estão descritos desde o primeiro estudo publicado por Beck et al.1 O tromboembolismo venoso é a manifestação mais comum ocorrendo em cerca de 40% dos casos1,17e em média nos primeiros dois anos após o início dos sintomas inflamatórios. A trombose arterial pode ocorrer, embora não seja tão frequente.16 Num estudo realizado por Alcedo et al verificou-se que em 54% dos eventos trombóticos não se identificou fator desencadeante. Ainda não é totalmente compreendida a via fisiopatológica subjacente a estes eventos, mas parece estar relacionada com inflamação, ativação endotelial e desregulação do sistema imune. No mesmo estudo parece existir uma relação entre estes eventos e as manifestações cardíacas da doença (miocardite, pericardite, derrame pericárdico).26
Outros achados menos comuns incluem manifestações gastrointestinais, como hepatoesplenomegalia,17 microabcessos hepáticos e colite,5,16manifestações genito-urinárias, que incluem orquiepididimite, prostatite5,16,17e, menos frequentemente, envolvimento renal com proteinúria ou desenvolvimento de insuficiência renal.10,16Na série de casos de Van der Made et al, foram observadas ainda múltiplas manifestações do sistema nervoso central (SNC) em doentes com VEXAS, desde cefaleias, polineuropatias, acidentes vasculares cerebrais, e ainda um caso de meningite asséptica.16Estão ainda descritos casos de disfunção vestibular e surdez.3,10
A Tabela 1 resume as principais características clínicas abordadas previamente e a sua prevalência.
EXAMES COMPLEMENTARES DE DIAGNÓSTICO
Tendo em conta a sintomatologia variada, muitas vezes o diagnóstico é tardio, podendo ocorrer em média entre 3 a 10 anos desde o início dos sintomas.6,12O diagnóstico de VEXAS é baseado na interpretação e combinação das manifestações clínicas anteriormente referidas e achados nos exames complementares de diagnóstico.
AVALIAÇÃO ANALÍTICA
Além das já referidas anemia macrocítica e trombocitopenia, podemos encontrar leucopenia, linfopenia e monocitopenia.15 Outro achado prevalente é a elevação de parâmetros inflamatórios, nomeadamente da proteína C reativa,1,7,12,17,24velocidade de sedimentação,1,7,12fator de necrose tumoral, IL-6 e IL-8, interferão, proteína 10 induzida pelo interferão1 e ferritina.16 Alguns casos podem apresentar anticorpo antinuclear ou anticoagulante lúpico positivo,13 aumento dos níveis de fator de von Willebrand ou do fator VIII.26
ESFREGAÇO DE SANGUE PERIFÉRICO
Apresenta-se tipicamente com macrocitose ligeira-moderada. Podemos também encontrar precursores de granulócitos imaturos, neutrófilos com vacúolos citoplasmáticos, hipogranulares e hipossegmentados, alguns com morfologia tipo Pelger-Huet, e monócitos vacuolizados.15
MIELOGRAMA
Os vacúolos citoplasmáticos nos precursores eritróides e mieloides são uma manifestação chave na síndrome VEXAS,1,12,25,27contudo estes achados são também observados no alcoolismo, toxicidade por zinco ou défice de cobre.3,21A ausência de vacúolos não deve ser critério de exclusão.16,28No mielograma é comum a presença de hipercelularidade com hiperplasia de granulócitos,3,7,12,16dispoiese ou atipia em megacariócitos e precursores eritroides e mieloides.15Nos doentes com SMD associado é característico o aumento do rácio mieloide/eritroide, hiperplasia mieloide, hipoplasia eritroide e baixas contagens de blastos.15 A citometria de fluxo pode revelar diminuição do número de precursores de células B, inversão do rácio CD4:CD8, expressão aberrante de CD56 e aumento das células T CD57+.15Não existem achados patognomónicos associados à síndrome VEXAS e nem todos os doentes têm alterações na avaliação medular.29
GENÉTICA
O diagnóstico definitivo da síndrome VEXAS é feito através da identificação da mutação UBA1 pelo método de Sanger,30 contudo a análise é demorada e não reconhece variações.31
TOMOGRAFIA COMPUTORIZADA DE TÓRAX
A realização de tomografia computorizada de tórax deve ser equacionada em doentes com queixas respiratórias. Os achados imagiológicos mais frequentemente descritos são a opacidade em vidro despolido, com ou sem consolidação, e a presença de nódulos pulmonares. Também pode ocorrer derrame pleural, normalmente pequeno, unilateral e predominantemente do lado direito.20 Até à data não se verificou evolução para fibrose pulmonar.20,32
LAVADO BRONCO-ALVEOLAR
O material obtido através de lavado bronco-alveolar é tipicamente exsudativo, de acordo com os critérios de Light, com predomínio de macrófagos e ainda com alguns linfócitos, neutrófilos e eosinófilos.20
Assim, tendo em conta as manifestações clínicas descritas e exames complementares enunciados, devemos pensar no diagnóstico de VEXAS perante um doente do sexo masculino, com idade superior a 50 anos, sintomas inflamatórios sistémicos, envolvimento cutâneo e disfunção hematológica. Num estudo que analisou 92 doentes com PR, um algoritmo baseado no sexo masculino, volume globular médio >100 fL, e plaquetas < 200x10^9/L, permitiu distinguir entre PR clássica e PR associada a VEXAS com uma sensibilidade de 100% e especificidade 96%.6
PROGNÓSTICO
Dos estudos realizados objetivou-se uma taxa de mortalidade de 40%,1 e uma taxa de sobrevida global aos 5 anos de 60%.17 A principal causa de morte está associada a citopenias, infeções secundárias ao estado de imunossupressão decorrente da terapêutica e disfunção hematológica, bem como a reações adversas graves ao próprio tratamento. Já foram identificados alguns fatores de mau prognóstico, nomeadamente o tipo de mutação (p.Met41Val), dependência de transfusões, envolvimento pulmonar, gastrointestinal e a identificação de gânglios mediastínicos.24
TRATAMENTO
A apresentação clínica da síndrome VEXAS, que reúne características sobrepostas entre síndrome inflamatória e disfunção hematológica, representa um desafio na adequada gestão destes doentes, necessitando frequentemente de uma perspetiva multidisciplinar.3 Esta patologia apresenta elevada morbimortalidade devido à persistência e refratariedade dos sintomas e à progressão hematológica.12,15
Atualmente a principal estratégia terapêutica assenta na corticoterapia em alta dose, geralmente com doses equivalentes de prednisolona superior a 20 mg diários.33Os efeitos adversos destes fármacos não podem ser ignorados e a recaída sintomática tem sido praticamente transversal a todos os doentes em que se faz uma tentativa de redução da dose de corticoide.33,34Várias publicações têm relatado falência terapêutica a diversas combinações de imunossupressores, reforçando a dificuldade na abordagem desta patologia.15,33,35Diversos estudos têm analisado um vasto número de opções terapêuticas, demonstrando maior tempo sem recaída sintomática com o uso de Azacitadina, no entanto com melhoria marginal das citopenias das diferentes linhagens hematológicas.33,36Alguns inibidores da JAK-1/2, principalmente o ruxolitinib, demonstraram resultados positivos com controlo sintomático e redução da corticoterapia.34Os inibidores da IL-6 como o tocilizumab revelaram algum potencial, com capacidade moderada de controlar a inflamação sistémica.12Outra estratégia terapêutica em avaliação é o transplante autólogo de células estaminais, com alguns casos clínicos descritos de controlo sintomático e remissão hematológica mantida pelo menos ao longo de 38 meses de follow-up.3,35Tendo em conta as complicações e efeitos adversos, este tratamento deverá ser equacionado apenas em casos refratários, com citopenias graves e irreversíveis, encontrando-se ainda por identificar o subgrupo de doentes que realmente beneficie desta intervenção.3,35O transplante autólogo de células estaminais parece ser a única medida disponível com intuito curativo.3 Além do tratamento dirigido, algumas medidas gerais revelam utilidade clínica, tais como a vacinação e profilaxia microbiana em doentes linfopénicos e avaliação do benefício de hipocoagulação, pelo elevado risco trombótico desta síndrome.3
CONCLUSÃO E PERSPETIVAS PARA O FUTURO
A identificação da síndrome VEXAS reforça a conexão entre patologias hematopoiéticas clonais e inflamação sistémica, podendo esta ser a primeira doença identificada de um novo grupo nosológico de doenças hematoinflamatórias.33A descoberta desta patologia poderá abrir a porta para a descoberta de novas doenças que resultem de mutações somáticas em células hematopoiéticas com consequente síndrome inflamatória e risco de transformação mielodisplásica, mieloproliferativa ou linfoproliferativa.33 Tendo em conta a sua identificação recente, a maioria dos estudos disponíveis apresentam limitações relacionadas com a metodologia e a curta duração do seguimento, encontrando-se ainda por definir as melhores estratégias terapêuticas. Estudos prospetivos com novos regimes terapêuticos deverão ser realizados com o objetivo de avaliar a sua eficácia e identificar subpopulações de doentes que beneficiem de diferentes regimes terapêuticos.15,33Com o avanço do conhecimento e crescente identificação de doentes com síndrome VEXAS, os dados sugerem que a sua prevalência será superior ao inicialmente estimado. Esta revisão pretende divulgar a evidencia científica existente e aumentar o grau de suspeição clínica da comunidade médica.

