










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkJornal Português de Gastrenterologia
versão impressa ISSN 0872-8178
J Port Gastrenterol. vol.19 no.4 Lisboa jul. 2012
Colangite esclerosante primária de pequenos ductos
Small duct primary sclerosing cholangitis
Gilberto Coutoa,∗, Pedro Barreiroa, Miguel Bispoa, Ana Rita Herculanoa, Sara Turpinb e Leopoldo Matosa
a Serviço de Gastrenterologia, Centro Hospitalar de Lisboa Ocidental, Lisboa, Portugal
b Serviço de Anatomia Patológica, Centro Hospitalar de Lisboa Ocidental, Lisboa, Portugal
*Autor para correspondência
Resumo
Apresentamos o caso de uma mulher de 18 anos com doença de Crohn do cólon agudizada, com envolvimento gastroduodenal, anasarca, pioderma gangrenoso e colestase sem icterícia. O colangiograma revelou-se normal e a biopsia hepática foi sugestiva de colangite esclerosante primária, fazendo por isso o diagnóstico da variante de pequenos ductos.
Revemos a literatura sobre esta doença muito rara que, tudo indica, se trata de uma entidade diferente da colangite esclerosante de grandes ductos, nomeadamente com melhor prognóstico.
Palavras-chave Colangite esclerosante primária; Colangite esclerosante primária de pequenos ductos; Doença de Crohn
Abstract
We report the clinical case of an 18-year-old woman admitted with Crohns disease with colitis and gastro duodenal involvement, anasarca, pyoderma gangrenosum and a pattern of anicteric cholestasis. The MR-cholangiogram was normal and liver biopsy suggested the presence of primary sclerosing cholangitis, thus making the diagnosis of small duct primary sclerosing cholangitis.
We review the state-of-the-art on this very rare disease, which seems a different clinical entity than large duct primary sclerosing cholangitis, and with a better prognosis.
Keywords Primary sclerosing cholangitis; Small duct primary sclerosing cholangitis; Crohns disease
Introdução
A colangite esclerosante primária (CEP) é uma manifestação extra-intestinal da doença inflamatória intestinal idiopática, em geral do cólon, relativamente rara. Não tem tratamento médico e o seu prognóstico é reservado, a menos que o doente seja submetido a um transplante hepático1-3.
Há uma variante de CEP, de pequenos ductos (CEP-PD), que pressupõe colangiograma normal, sendo por isso diagnosticada por biopsia hepática. Trata-se, segundo parece atualmente, de uma entidade própria, com melhor prognóstico que a CEP de grandes ductos4,5.
Apresentamos o caso de uma mulher de 18 anos com doença de Crohn do cólon agudizada e colestase sem icterícia com colangiograma normal. A biopsia hepática foi sugestiva de CEP, fazendo-se por isso o diagnóstico da variante de pequenos ductos. Revemos o estado atual do conhecimento sobre esta doença.
Caso clínico
Relata-se o caso clínico de uma mulher de 18 anos, caucasiana, natural do Brasil, com o diagnóstico de colite de Crohn aos 8 anos. Sem seguimento nem tratamento posteriores, refere ter estado assintomática até ao verão de 2009.
Nessa ocasião, iniciou quadro de diarreia, 4-6 dejeções por dia, por vezes com sangue, desconforto abdominal difuso e episódios de vómitos, por vezes hematemeses de sangue digerido em pequena quantidade, emagrecimento não quantificado, edemas generalizados e úlceras cutâneas em número e extensão crescentes.
Dado o agravamento progressivo das queixas recorreu ao hospital, em setembro de 2009, sendo internada no nosso serviço.
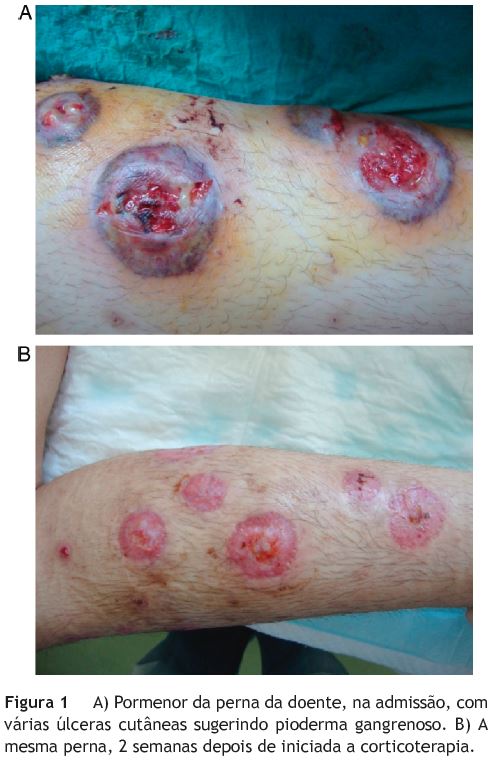
No exame físico salientavam-se palidez muco-cutânea, anasarca e úlceras cutâneas violáceas dolorosas com centro necrótico-purulento, em maior número nos membros inferiores, sugerindo pioderma gangrenoso (fig. 1).
Analiticamente destacavam-se anemia normocítica normocrómica, com hemoglobina de 8 g/dL, leucograma com 10.800 células/mL com 83% de neutrófilos e PCR 4,7 mg/dL. A amilase e a lipase eram normais. As serologias virais para o vírus da imunodeficiência humana, o vírus da hepatite B e o vírus da hepatite C eram negativas. As enzimas hepáticas, a albumina e o tempo de protrombina revelavam: colestase crónica e hipoalbuminemia graves com transaminases normais (tabela 1).
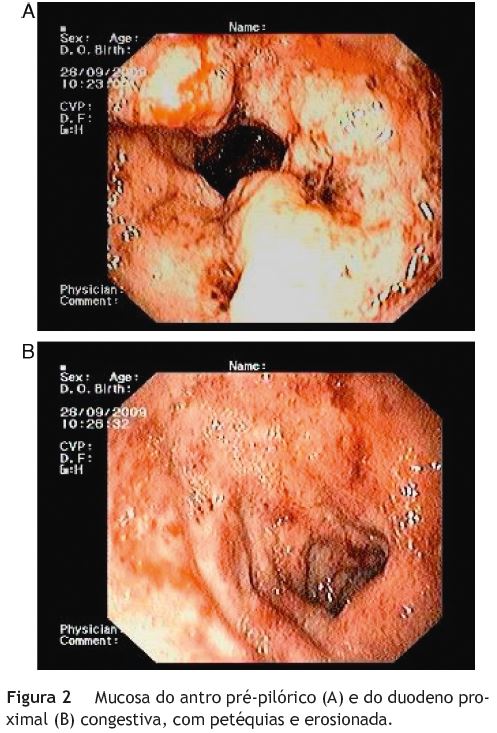
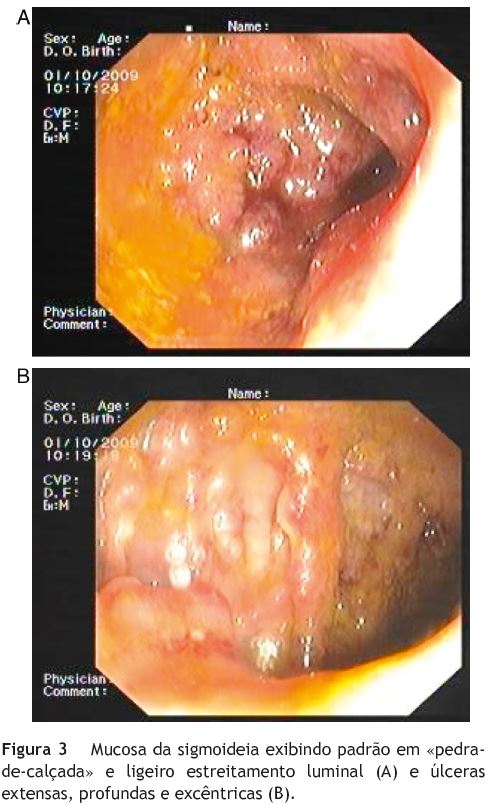
Fizeram-se endoscopia digestiva alta e fibrosigmoidoscopia. A endoscopia mostrou mucosa do antro e do duodeno proximal muito congestiva e irregular, com múltiplas erosões e friável (fig. 2), levantando a suspeita de doença de Crohn gastro-duodenal. A fibrosigmoidoscopia mostrou úlceras extensas, profundas e excêntricas da mucosa do cólon, a par de mucosa congestiva e sangrante, exibindo padrão em «pedra-de-calçada» (fig. 3). As biopsias de ambos os exames endoscópicos foram, no entanto, inconclusivas, não se encontrando granulomas, nem Helicobacter pylori nas biopsias gástricas, nem CMV nas biopsias do cólon.
A TAC abdómino-pélvica revelou fina lâmina de ascite, edema da parede abdominal e ausência de abcessos intraabdominais. Assim, após culturas dos líquidos biológicos e zaragatoas das úlceras cutâneas, que vieram negativas, iniciou-se prednisolona (50 mg/d), metronidazol e ciprofloxacina. Estava ainda medicada com mesalazina, ferro, albumina e esomeprazol.
No entanto, a doente desenvolveu síndrome de dificuldade respiratória do adulto com necessidade de ventilação mecânica em unidade de cuidados intensivos (UCI), onde esteve uma semana sob alimentação parentérica total e iniciou isoniazida (dada corticoterapia). Nessa ocasião houve melhoria clínica progressiva mas agravamento da elevação das enzimas hepáticas (tabela 1), embora sem encefalopatia hepática e com tempo de protrombina, bilirrubina total e albumina dentro do normal. Suspendeu-se a isoniazida e a alimentação parentérica total após a transferência da UCI para o nosso serviço.
Por ocasião da alta a doente encontrava-se assintomática, sem edemas e com as úlceras cutâneas em avançado estado de cicatrização (fig. 1b). Os valores analíticos constam na tabela 1. A doente teve alta medicada com ácido ursodesoxicólico (AUDC) 1.000 mg/d, mesalazina oral e retal e prednisolona oral. Manteve ainda, durante um período, esomeprazol e ferro, e iniciou azatioprina em dose baixa, que se foi aumentando em ambulatório.
Repetiu, alguns meses após a alta, a endoscopia, já sem alterações, e a colonoscopia, que mostrou íleon normal e pseudopólipos dispersos em mucosa cólica de resto íntegra (biopsias com «inflamação crónica inespecífica»).
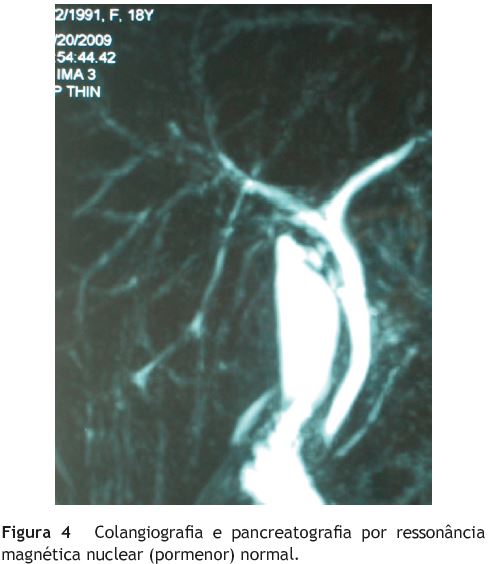
Realizou colangio-pancreatografia por ressonância magnética nuclear (CPRMN), que não mostrou alterações (fig. 4). Ainda para esclarecimento das alterações hepáticas, pesquisaram-se os auto-anticorpos pANCA, anti-nuclear, anti-músculo liso, anti-mitocondrial e anti-LKM. O pANCA PR3 foi o único positivo. A Ig G4 era normal e os métodos de imagem mostraram sempre veia porta permeável.
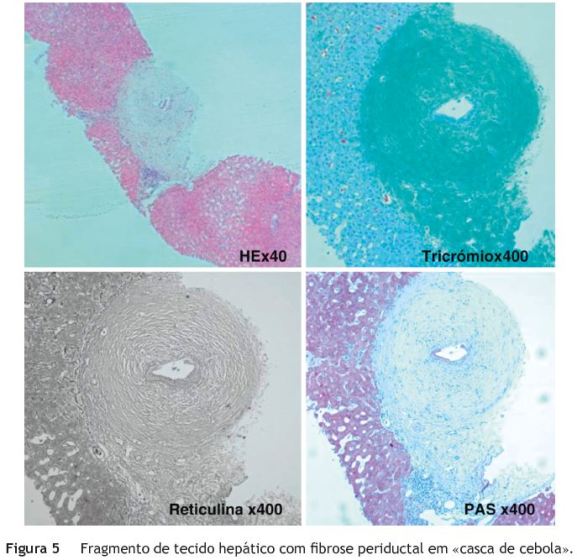
Por manter enzimas hepáticas elevadas, com predomínio do padrão colestático, realizou-se biopsia hepática percutânea que revelou aspetos sugestivos de CEP, com a característica lesão de fibrose periductal em «casca de cebola» (fig. 5).
Encontra-se assintomática 9 meses depois da alta, medicada com azatioprina, mesalazina e AUDC, a que adere irregularmente.
Discussão
Apresentámos um caso de doença de Crohn do cólon agudizada, com envolvimento gastroduodenal invulgar. Esta foi uma das razões para a introdução precoce de azatioprina. Diagnosticaram-se ainda, na admissão, pioderma gangrenoso, com excelente resposta à corticoterapia, e colestase sem icterícia sugerindo a hipótese de CEP. Durante o internamento, houve agravamento da colestase e elevação das aminotransferases por provável «toxicidade» da alimentação parentérica total e da isoniazida. Por isso se diferiu a biopsia hepática durante alguns meses, sabendo-se que o colangiograma era normal.
Mas, a propósito deste caso, privilegiámos nesta discussão uma revisão da CEP-PD, dada a sua raridade.
Epidemiologia
A CEP tem uma prevalência e incidência anual estimadas de 3,85-8,5 e 0,41-1,3 casos por 100.000 habitantes, respetivamente3,6,7. A CEP-PD é uma doença ainda mais rara: descrita por Wee e Ludwig há cerca de 20 anos8,9, só um pequeno número de casos foi até agora relatado, em parte - certamente - por subnotificação1.
A maioria dos casos de CEP e CEP-PD associa-se à doença inflamatória intestinal idiopática do cólon, embora se saiba que menos de 5% dos doentes com doença inflamatória intestinal têm CEP8. A CEP-PD representa apenas 5,8-11% do total de casos de CEP4,10,11.
A CEP-PD, tal como a CEP, é uma doença tipicamente dos homens com colite ulcerosa. Algumas séries demonstraram, no entanto, proporções relativamente maiores de colite de Crohn e de mulheres na CEP-PD do que na CEP4,5. Tal como mais casos de síndromes de sobreposição, nomeadamente com a hepatite auto-imune, presente em 10-27% dos doentes com CEP-PD7,12.
Clínica e diagnóstico
A presença de colestase, crónica, especialmente em doente anictérica com colite de Crohn é muito sugestiva de CEP. A CPRMN normal obriga a biopsia hepática para confirmar ou não a presença de CEP-PD, diagnóstico confirmado nesta doente.
A CPRMN tem sido, cada vez mais, considerada o exame de primeira linha perante a suspeita de CEP, desde que não seja previsível uma atitude terapêutica1. Nos colangiogramas normais nem sempre a biopsia hepática, nomeadamente a percutânea, é esclarecedora, por dificuldades de amostragem e baixa especificidade dos achados. A integração da clínica e do laboratório com os achados da CPRMN (ou CPRE) e da biopsia hepática é por isso fundamental.
Tratamento
Na CEP avançada, a única opção terapêutica é o transplante hepático, com 85-90% de sobrevida aos 5 anos13 e, em geral, melhoria dos sintomas da doença inflamatória intestinal3. Nenhum medicamento altera, contudo, a história natural da CEP. O AUDC parece melhorar a colestase bioquímica mas não melhora os sintomas, não influencia a progressão da doença e não reduz a mortalidade1-3,14-16. Resta confirmar se poderá ser usado como agente quimioprofilático do colangiocarcinoma e do carcinoma do cólon e do reto, como foi demonstrado em doentes com colite ulcerosa17.
Na nossa doente, esta poderá ser, definitivamente, a única razão para manter o AUDC, introduzido empiricamente antes do diagnóstico definitivo, e cuja manutenção deverá ser repensada.
Prognóstico
A CEP-PD tem melhor prognóstico que a CEP, iniciando-se ambas por volta da mesma idade e sem que a primeira evolua para a segunda na maioria dos casos, o que sugere tratarem-se de entidades diferentes. A CEP-PD pode, no entanto, evoluir para CEP em 12 e 23% dos casos após 5 e 7 anos de seguimento, respetivamente4,5,18. A CPRMN é uma forma simples de monitorizar esta progressão, embora os intervalos de vigilância e o seu custo-eficácia não estejam definidos.
A CEP-PD, sem a progressão para lesões de grandes ductos, não tem risco de colangiocarcinoma4,5,10,18. Já na CEP de grandes ductos ocorreram, nos mesmos estudos, 11-12% de colangiocarcinomas, no mesmo período de seguimento4,5,10. Nestas séries, a percentagem de óbitos e transplantados hepáticos foi de 9-23% nos doentes com CEP-PD e 42-50% nos doentes com CEP. A doença reapareceu no fígado transplantado em 2 de 8 transplantados com CEP-PD: após 9 anos num caso e 13 anos no outro5.
O prognóstico da CEP-PD não parece ser diferente nos doentes sintomáticos e assintomáticos aquando do diagnóstico4 ou com e sem doença inflamatória intestinal5,14. Pensa-se que, à semelhança da CEP, a colectomia não parece influenciar o aparecimento e a progressão da doença colestática, a menos que o doente seja transplantado, situação em que a colectomia se associa a menos recidivas de CEP no enxerto1,2.
Finalmente, como a doente se encontra assintomática, o relevo do diagnóstico de CEP-PD centra-se na vigilância: da função hepática e da eventual progressão para a CEP de grandes ductos - antecipando o risco acrescido de colangiocarcinoma - e do carcinoma do cólon e do reto.
Bibliografia
1. Heathcote J. Sclerosing cholangitis in 2009. Em: Arroyo V, Abraldes JG, Ginés P, Sánchez-Tapias JM, Forns X, Bataller R, Rodés J, editores Treatment of liver diseases. Barcelona: Ars Medica; 2009. p. 291-300. [ Links ]
2. Macedo G. Colangite esclerosante primária. Em: Areias J, editor. Tratado de hepatologia. Lisboa: Permanyer Portugal; 2006. p. 343-58. [ Links ]
3. Lee YM, Kaplan MM. Management of primary sclerosing cholangitis. Am J Gastroenterol. 2002;97:528-34. [ Links ]
4. Björnsson E, Boberg KM, Cullen S, Fleming K, Clausen OP, Fausa O, et al. Patients with small duct primary sclerosing cholangitis have a favourable long term prognosis. Gut. 2002;51:731-5. [ Links ]
5. Björnsson E, Olsson R, Bergquist A, Lindgren S, Braden B, Chapman RW, et al. The natural history of small-duct primary sclerosing cholangitis. Gastroenterology. 2008;134:975-80. [ Links ]
6. Card TR, Solaymani-Dodaran M, West J. Incidence and mortality of primary sclerosing cholangitis in the UK: a population-based cohort study. J Hepatol. 2008;48:939-44. [ Links ]
7. Kaplan GG, Laupland KB, Butzner D, Urbanski SJ, Lee SS. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-basedd analysis. Am J Gastroenterol. 2007;102:1042-9. [ Links ]
8. Wee A, Ludwig J. Pericholangitis in chronic ulcerative colitis: primary sclerosing cholangitis of the small bile ducts. Ann Intern Med. 1985;102:581-7. [ Links ]
9. Ludwig J. Small duct primary sclerosing cholangitis. Semin Liver Dis. 1991;11:11-7. [ Links ]
10. Angulo P, Maor-Kendler Y, Lindor KD. Small-duct primary sclerosing cholangitis: a long-term follow-up study. Hepatology. 2002;35:1494-500. [ Links ]
11. Angulo P, Maor-Kendler Y, Donlinger JJ, Lindor KD. Small-duct primary sclerosing cholangitis: prevalence and natural history. Gastroenterology. 2000;118:A902. [ Links ]
12. Olsson R, Glaumann H, Almer S, Broomé U, Lebrun B, Bergquist A, et al. High prevalencee of small-duct primary sclerosing cholangitis among patients with overlapping autoimmune hepatitis and primary sclerosing cholangitis. Eur J Intern Med. 2009;20:190-6. [ Links ]
13. Gow PJ, Chapman RW. Liver transplantation for primary sclerosing cholangitis. Liver. 2000;20:97-103. [ Links ]
14. Charatcharoenwitthaya P, Angulo P, Enders FB, Lindor KD. Impact of inflammatory bowel disease and ursodeoxycholic acid therapy on small-duct primary sclerosing cholangitis. Hepatology. 2008;47:133-42. [ Links ]
15. Smith T, Befeler AS. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Curr Gastroenterol Rep. 2007;9:54-9. [ Links ]
16. Vacca M, Krawczyk M, Petruzzelli M, Sasso RC, Van Erpecum KJ, Palasciano G, et al. Current treatments of primary sclerosing cholangitis. Curr Med Chem. 2007;14:1094-2081. [ Links ]
17. Pardi DS, Loftus Jr EV, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889-93. [ Links ]
18. Broomé U, Glaumann H, Lindstöm E, Lööf L, Almer S, Prytz H, et al. Natural history and outcome in 32 swedish patients with small duct primary sclerosing cholangitis (PSC). J Hepatol. 2002;36:586-9. [ Links ]
Conflito de interesses
Os autores declaram não haver conflito de interesses.
*Autor para correspondência
Correio eletrónico: gpcouto@sapo.pt (G. Couto).
Agradecimentos
À Dra. Sância Ramos, pelo apoio dado.
Recebido a 29 de julho de 2010; aceite a 18 de novembro de 2010
