










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkJornal Português de Gastrenterologia
versão impressa ISSN 0872-8178
J Port Gastrenterol. vol.20 no.3 Lisboa maio 2013
https://doi.org/10.1016/j.jpg.2012.07.005
CLINICAL CASE
Síndrome de Klippel-Trenaunay-Weber - Hemorragia gastrointestinal em doente jovem
Klippel-Trenaunay-Weber Syndrome - Gastrointestinal hemorrhage in a young patient
Carlos Fernandes∗, Luís Alberto, Rolando Pinho, Ricardo Veloso, Teresa Pinto-Pais, João Carvalho e José Fraga
Serviço de Gastrenterologia, Centro Hospitalar Vila Nova de Gaia/Espinho, Vila Nova de Gaia, Portugal
*Autor para correspondência
RESUMO
A síndrome de Klippel-Trenaunay-Weber é uma patologia rara que se manifesta por malformações capilares cutâneas, anomalias venosas e/ou linfáticas, hipertrofia óssea e/ou de tecidos moles associadas a malformações arteriovenosas. O envolvimento do sistema gastrointestinal ocorre em cerca de 1/5 dos pacientes e manifesta-se habitualmente por hemorragia digestiva baixa. O tratamento é predominantemente sintomático. Os autores apresentam o caso de uma jovem de 19 anos, com diagnóstico de síndrome de Klippel-Trenaunay-Webber referenciada à consulta de Proctologia por episódios de retorragias episódicas desde a infância. Concomitantemente, apresentava insuficiência venosa no membro inferior direito, hemangiomas capilares cutâneos em ambos os membros inferiores e hipertrofia do membro inferior esquerdo. Ao exame objetivo observaram-se hemorroidas grau ii congestivas, mas não friáveis. Endoscopicamente, o reto e sigmoide distal apresentavam vários cordões varicosos, de aspeto polipóide, recobertos de mucosa normal. Iniciou terapêutica com laxantes e fibras alimentares com melhoria dos sintomas.
Palavras-Chave: Klippel-Trenaunay-Weber Syndrome; Hemorragia Gastrointestinal; Proctologia
ABSTRACT
Klippel-Trenaunay-Weber syndrome is a rare pathology characterized by the occurrence of capillary cutaneous malformations, venous and/or lymphatic anomalies, bony and/or soft tissue hypertrophy associated with arteriovenous malformations. Gastrointestinal involvement occurs in about 1/5 of affected patients and usually presents as lower gastrointestinal bleeding. Treatment is mainly symptomatic. The authors present a clinical case of a 19 year-old woman with known Klippel-Trenaunay-Weber syndrome who was referred to our Proctology outpatient clinic due to sporadic rectal bleeding since early childhood. She had venous insufficiency of her right lower limb, capillary cutaneous hemangiomas in both lower limbs and hypertrophy of the left lower limb. Proctologic examination revealed grade II haemorrhoids, which were congested but not friable. On flexible sigmoidoscopy, several varicose veins with polypoid appearance and endoscopically normal mucosa were apparent. Management with laxatives and dietary fiber increase was initiated with symptomatic improvement.
Keywords: Klippel-Trenaunay-Weber Syndrome; Gastrointestinal hemorrhage; Proctology
Introdução
A síndrome de Klippel-Trenaunay-Weber (SKTW)é uma patologia rara que se manifesta tipicamente por malformações capilares cutâneas, anomalias venosas e/ou linfáticas, hipertrofia óssea e/ou dos tecidos moles associadas a malformações arteriovenosas. O envolvimento do sistema gastrointestinal, através da presença de hemangiomas e veias varicosas, ocorre em cerca de 20% dos pacientes sendo na sua maioria assintomático. Quando sintomático, manifesta-se habitualmente por hemorragia digestiva baixa, nomeadamente hematoquézias ou retorragias. A abordagem terapêutica depende do local de envolvimento do sistema gastrointestinal e da severidade da sintomatologia.
Caso clínico
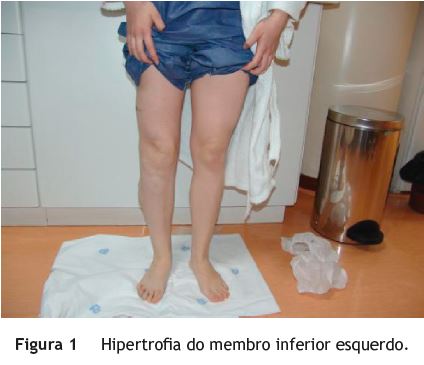
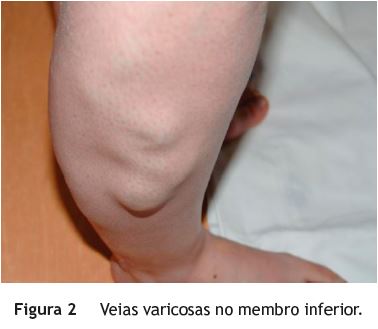
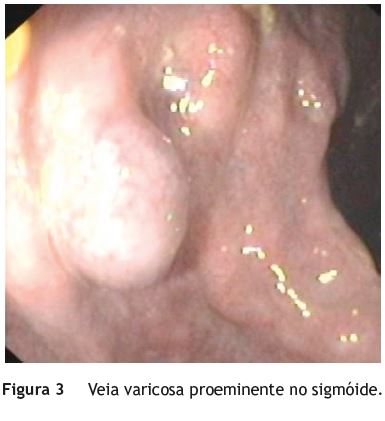
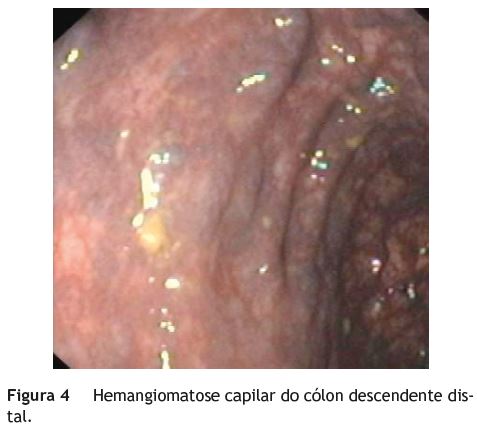
Jovem, sexo feminino, 19 anos, com diagnóstico de SKTW referenciada à consulta de Proctologia por retorragias episódicas desde a infância. A doente referia retorragias espontâneas e associadas a esforço defecatório, frequentes (2-3 vezes/dia), autolimitadas, sem repercussão hemodinâmica ou analítica. Sem outra sintomatologia nomeadamente alterações do trânsito intestinal, dor abdominal ou perianal. No contexto da síndrome apresentava ainda hipertrofia do membro inferior esquerdo (fig. 1), disfunção venosa no membro inferior direito (fig. 2) (com episódios prévios de trombose venosa) e hemangiomas capilares cutâneos em ambos os membros inferiores. No exame proctológico a destacar marisca anal na comissura posterior e hemorroidas grau ii congestivas, mas não friáveis. Na colonoscopia total observou-se, no reto e sigmoide distal, vários cordões varicosos proeminentes, assumindo por vezes aspeto polipoide, recobertos de mucosa normal (fig. 3). No sigmoide proximal constatou-se rede venosa submucosa exuberante, aracniforme, muito ramificada, recoberta de mucosa normal - aspeto sugestivo de hemangiomatose capilar (fig. 4). Angio-ressonância magnética abdomino-pélvica e endoscopia digestiva alta sem alterações de relevo. Na videocápsula endoscópica observou-se mucosa congestionada circunferencialmente e com aspeto polipóide no jejuno proximal e procidência recoberta de mucosa normal (8-10mm) no jejuno distal.
Durante seguimento em ambulatório, observou-se uma paulatina diminuição do valor de hemoglobina no contexto de retorragias.
Iniciou tratamento conservador com hidratação abundante, alimentação rica em fibras e laxantes em SOS no sentido de regularizar trânsito intestinal e diminuir esforço defecatório. Por anemia ferropénica iniciou também suplementação com ferro oral. Desde então com melhoria progressiva, mantendo apenas raros episódios de retorragias. Mantém seguimento na consulta de proctologia e encontra-se alertada para sinais de alarme.
Discussão
Em 1900, Klippel e Trenaunay descreveram uma síndrome de alterações do sistema vascular que incluía a tríade de malformação capilar cutânea, anomalias venosas e/ou linfáticas e hipertrofia óssea/tecidos moles. Posteriormente, Parkes e Weber identificaram pacientes com a tríade anterior associada a malformações arteriovenosas --- SKTW.
Atualmente, considera-se uma patologia congénita, com manifestação esporádica. No entanto, estão descritos eventuais casos de transmissão Mendeliana, em especial de acordo com um padrão de transmissão autossómico paradominante1. Segundo esta última hipótese, os homozigotos para o gene envolvido seriam inviáveis e os heterozigotos saudáveis; através da perda de heterozigotia durante a embriogénese (ou seja, por mutação somática) existiria manifestação fenotípica da doença.
O gene AGGF1, que codifica um fator de crescimento angiogénico, tem sido recorrentemente envolvido2. No entanto, é ainda necessária maior evidência científica.
A etiopatogenia associada a esta síndrome não se encontra, igualmente, esclarecida. No entanto, alterações na embriogénese dos vasos linfáticos e vasculares, mais concretamente da mesoderme destas estruturas, surgem como hipótese mais provável3.
O SKTW tem distribuição equitativa a nível geográfico, racial e de género4.
Para efeitos de diagnóstico basta a presença de 2 das anomalias atrás referidas. No entanto, a totalidade das alterações estão presentes na maioria dos doentes, sendo a malformação capilar cutânea a anomalia mais frequente. Estima-se que as alterações venosas/linfáticas (incluindo as malformações arteriovenosas) atinjam cerca de 70% e a hipertrofia dos membros apenas 65% dos casos. As manifestações clínicas desta patologia surgem, habitualmente, à nascença ou durante a infância.
O local de apresentação mais frequente é uma das extremidades corporais, nomeadamente o membro inferior. No entanto, estão descritos casos de envolvimento de mais do que uma extremidade, assim como o envolvimento do tronco, pescoço ou cabeça.
As alterações capilares mais comuns são os hemangiomas. Geralmente superficiais, estas estruturas podem atingir igualmente camadas mais profundas e órgãos viscerais. Quando de maiores dimensões os hemangiomas podem condicionar sequestro de plaquetas dando origem à síndrome de Kasabach-Merrit - coagulopatia de consumo com prognóstico reservado5.
As alterações varicosas são principalmente evidentes nas porções distais dos membros (p. ex. pé e tornozelo) manifestando-se à nascença ou apenas após deambulação, aos 3 a 4 anos de idade. A presença de veias varicosas em órgãos viscerais, como bexiga, cólon ou pulmão, é também possível.
As malformações arteriovenosas podem-se localizar em diversos locais e são evidentes através de massas pulsáteis, frémitos ou sopros. Além disso, a hipertermia do membro envolvido e o sinal de Braham (bradicardia ao pressionar a artéria proximal à malformação) são também sugestivas desta anomalia.
A hipertrofia do membro afetado deve-se quer à hipertrofia do tecido ósseo quer à do tecido muscular. A alteração pode ser evidente à nascença, com tendência a se tornar mais evidente com o crescimento. Estão descritos casos de diferenças significativas no desenvolvimento, sendo as maiores discrepâncias associadas à presença de malformações arteriovenosas.
O envolvimento visceral do SKTW parece mais prevalente do que inicialmente pensado, estimando-se que envolva cerca de 20% dos pacientes6. As estruturas possivelmente envolvidas são diversas, tais como a pleura, baço, fígado, ou sistema gastrointestinal (GI). Este envolvimento, como previamente referido, deve-se essencialmente à presença de hemangiomas e veias varicosas.
No sistema gastrointestinal, as áreas frequentemente afetadas são o sigmoide e reto, e em menor frequência o jejuno e esofágo. O envolvimento preferencial do reto e sigmoide dever-se-á a malformações nas veias popliteias e femurais superficiais que condicionam alterações no fluxo arterial assim como hipertensão na drenagem venosa do reto e sigmoide. Neste contexto, ocorrem dilatações venosas, veias varicosas, que pelas suas dimensões e pelo traumatismo provocado pelas fezes condicionam hemorragia7. No entanto, este processo não justifica os hemangiomas capilares e as alterações vasculares presentes no jejuno ou no esófago.
O envolvimento do aparelho gastrointestinal não condiciona, habitualmente, qualquer tipo de manifestação. No entanto, quando evidente, a manifestação clínica mais comum é a hemorragia, principalmente através de hematoquézias e/ou retorragias. O espetro da hemorragia GI é amplo, variando de hemorragia gastrointestinal oculta em doente assintomático até hemorragia digestiva maciça, com repercussão hemodinâmica. Geralmente é intermitente, sendo o primeiro episódio durante a infância e pode-se prolongar durante toda a vida.
Em doentes com as manifestações típicas o diagnóstico é evidente apenas com a história clínica e exame objetivo. No entanto, os meios auxiliares de diagnóstico podem revelarse úteis no diagnóstico e avaliação desta patologia.
Os estudos radiológicos, a ultrassonografia com ou sem doppler, a tomografia computorizada, a ressonância magnética e os estudos vasculares como a arteriografia e venografia são alguns dos métodos utilizados.
Na investigação do envolvimento do sistema gastrointestinal, nomeadamente coloretal, os métodos de imagem revelam-se de grande importância. Os estudos baritados podem evidenciar um estreitamento do lúmen facilmente distensível, fruto da protusão das varicosidades e dos hemangiomas na parede8. A tomografia computorizada9 e a ressonância magnética são métodos simples e não invasivos para avaliação de massas hemangiomatosas e sua extensão. O estudo vascular por angiografia é essencialmente útil para avaliação adequada da extensão das lesões, no préoperatório10.
A avaliação endoscópica assume também papel preponderante. É frequente a observação de vasos mucosos de grandes dimensões, hemangiomas mucosos, lesões angiomatosas volumosas da submucosa e nódulos vasculares principalmente no reto e sigmoide. No entanto, apesar da facilidade na identificação destas lesões distais, é mandatória a observação endoscópica de todo o sistema gastrointestinal, no sentido de excluir lesões metácronas. Apesar da raridade desta patologia devemos estar alerta, pois a realização de biopsias em estruturas vasculares poderá ser catastrófica, condicionando hemorragia de difícil controlo11.
Em situações de hemorragia gastrointestinal ativa a avaliação endoscópica é difícil e muitas vezes não tolerada pelo paciente. Nestes casos, graças à sua capacidade diagnóstica e acima de tudo à sua capacidade terapêutica a angiografia assume-se como gold-standard.
O tratamento dos doentes com SKTW baseia-se essencialmente no tratamento dos seus sintomas. O tratamento etiológico não é possível.
A abordagem dos hemangiomas depende das suas dimensões, manifestações ou complicações. A terapêutica com laser conduz a resultados animadores na sua vertente estética e em casos de ulceração.
O uso de compressão elástica nos membros inferiores é útil em casos de insuficiência venosa periférica e linfedema, na prevenção de celulite recorrente e da trombose venosa profunda e constitui ainda proteção para eventos traumáticos. A abordagem cirúrgica das alterações vasculares é considerada em casos selecionados. Dado o risco aumentado de tromboflebite, o uso de contracetivos orais está contraindicado nesta patologia.
Em situações de hipertrofia dos membros inferiores, a abordagem pode igualmente ser conservadora ou cirúrgica. Quando a discrepância entre membros é igual ou menor que 1,5 cm, o tratamento conservador com próteses e calçado ortopédico é a primeira escolha. Para discrepâncias maiores que 1,5 cm a abordagem é cirúrgica.
A abordagem do envolvimento colorretal pelo SKTW depende da sua extensão e da gravidade das perdas hemáticas. O tratamento com suplementação de ferro, a dieta rica em fibras e os laxantes podem ser considerados em doentes com hemorragias ligeiras, intermitentes, não debilitantes.
No entanto, com a progressão da doença, poderá ser necessária uma abordagem mais interventiva. A necessidade de suporte transfusional, a diminuição da qualidade de vida e a presença de hemorragia cataclísmica apresentam-se como critérios de mau prognóstico.
A abordagem endoscópica é ainda muito limitada, sendo apenas particularmente útil em lesões focais. A fotocoagulação com árgon-plasma12 e a utilização do laser NYG13 já foram descritas para o tratamento de lesões focais e/ou residuais, pós-cirurgia, com resultados animadores.
A angiografia com embolização dos segmentos vasculares anómalos surge como hipótese após identificação precisa de local de hemorragia. A abordagem cirúrgica pode também ser realizada em casos selecionados e passa pela retirada do segmento envolvido, privilegiando a preservação do esfíncter anal. A radioterapia tem surgido como nova modalidade terapêutica, sendo ainda necessária maior experiência14.
Bibliografia
1. Hofer T, Frank J, Itin PH. Klippel-Trenaunay syndrome in a monozygotic male twin: supportive evidence for the concept of paradominant inheritance. Eur J Dermatol. 2005;15: 341-3. [ Links ]
2. Hu Y, Li L, Seidelmann SB, Timur AA, Shen PH, Driscoll DJ, et al. Identification of association of common AGGF1 variants with susceptibility for Klippel-Trenaunay syndrome using the structure association program. Ann Hum Genet. 2008;72:636-43. [ Links ]
3. Baskerville PA, Ackroyd JS, Browse NL. The etiology of the Klippel-Trenaunay syndrome. Ann Surg. 1985;202:624-7. [ Links ]
4. Jacob AG, Driscoll DJ, Shaughnessy WJ, Stanson AW, Clay RP, Gloviczki P. Klippel-Trénaunay syndrome: spectrum and management. Mayo Clin Proc. 1998;73:28-36. [ Links ]
5. Dufau JP, le Tourneau A, Audouin J, Delmer A, Diebold J. Isolated diffuse hemangiomatosis of the spleen with Kasabach-Merrittlike syndrome. Histopathology. 1999;35:337-44. [ Links ]
6. Cha SH, Romeo MA, Neutze JA. Visceral manifestations of Klippel-Trenaunay syndrome. Radiographics. 2005;25:1694-7. [ Links ]
7. Kotze PG, Soares AV, Lima MC, Baldin-Junior A, Sartor MA, Bonardi RA. Síndrome de Klippel-Trenaunay: Uma causa rara de hemorragia digestiva baixa. Rev Bras Coloproct. 2002;22:109-12. [ Links ]
8. Ghahremani GG, Kangarloo H, Volberg F, Meyers MA. Diffuse cavernous hemangioma of the colon in the Klippel- Trenaunay syndrome. Radiology. 1976;118:673-8. [ Links ]
9. Yeoman LJ, Shaw D. Computerized tomography appearances of pelvic haemangioma involving the large bowel in childhood. Pediatr Radiol. 1989;19:414-6. [ Links ]
10. Cha SH, Romeo MA, Neutze JA. Visceral manifestations of Klippel-Trenaunay Syndrome. Radiographics. 2005;25:1694-7. [ Links ]
11. Gandolfi L, Rossi A, Stasi G, Tonti R. The Klippel-Trenaunay syndrome with colonic hemangioma. Gastrointest Endosc. 1987;33:442-5. [ Links ]
12. Azizkhan RG. Life-threatening hematochezia from a rectosigmoid vascular malformation in Klippel-Trenaunay syndrome: long-term palliation using an argon laser. J Pediatr Surg. 1991;26:1125-8. [ Links ]
13. Myers BM. Treatment of colonic bleeding in Klippel-Trenaunay syndrome with combined partial colectomy and ndoscopic laser. Dig Dis Sci. 1993;38:1351-3. [ Links ]
14. Grundfest-Broniatowski S, Carey WD, Sivak MV, Feldman B. Klippel-Trenaunay-Weber syndrome with visceral involvement and portal hypertension. Cleveland Clin Q. 1982;49:239-47. [ Links ]
Conflito de interesses
Os autores declaram não haver conflito de interesses.
*Autor para correspondência
Correio eletrónico: carlosdpfernandes@gmail.com (C. Fernandes).
Recebido a 8 de dezembro de 2011; aceite a 11 de março de 2012

{kind=link}