










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Pneumologia
versão impressa ISSN 0873-2159
Rev Port Pneumol v.16 n.1 Lisboa jan. 2010
CYP1A1 m1 and m2 polymorphisms: genetic susceptibility to lung cancer
Paula Mota1, David Silva Moura2, Maria Graça Vale3, Henriqueta Coimbra4, Lina Carvalho5, Fernando Regateiro6
1 PhD Student – Instituto de Genética
2 MA Student – Departamento de Zoologia
3 Full Professor – Faculdade de Ciências e Tecnologia
4 PhD M.D. Medical Genetics – Instituto de Genética
5 PhD M.D. Pathology – Instituto de Anatomia Patológica
6 PhD M. D. Medical Genetics – Instituto de Genética
Abstract
Lung cancer is considered an environment-related disease that develops as a consequence of exposure to mutagenic agents, namely those present in tobacco. The CYP1A1 gene codifies the phase I enzyme aryl hydrocarbon hydroxilase (AHH) belonging to the cytochrome P450 system that plays a major role in the bio-activation of tobacco procarcinogenes. Two CYP1A1 polymorphisms, m1 (T6235C transition) and m2 (A4889G transition), are associated with greater enzymatic activity and have been described as genetic susceptibility factors for lung cancer.
The aim of this study was to verify if this association holds true in blood samples of 175 lung cancer patients and 217 non-cancer patients from Portugal’s midlands region. The samples were studied by restriction fragment length polymorphism (RFLP) assay.
The allelic frequencies of the mutant alleles were 0.12 for allele C and 1.14 for allele G in the control population. The results were not statistically different from those alleles in the patient population. There was also no statistically significant difference in genotype distribution in lung cancer patients and controls even when combining high risk genotypes. In our control sample, as in other populations of different ethnic origin, both polymorphisms also seem to be in linkage disequilibrium. We conclude that in this sample of the Portuguese population, CYP1A1 m1 and m2 polymorphisms are too rare to be of clinical relevance, and do not seem to be associated with susceptibility to lung cancer.
Key-words: Lung cancer, smoking, cytochrome P450, CYP1A1, linkage disequilibrium.
Polimorfismos dos alelos m1 e m2 do gene CYP1A1: Susceptibilidade genética para o cancro do pulmão
Resumo
O cancro do pulmão é considerado uma doença relacionada com o meio ambiente, consequência da exposição a agentes mutagénicos, nomeadamente os presentes no fumo do tabaco. O gene CYP1A1 codifica a enzima aril hidrocarboneto hidroxilase (AHH), da fase I, do sistema multienzimático do citocromo P450, que desempenha uma função preponderante na bioactivação dos procarcinogénios do tabaco. Dois polimorfismos do CYP1A1, m1 (transição T6235C) e m2 (transição A4889G), estão associados a uma maior actividade enzimática, tendo sido referidos como factores genéticos de susceptibilidade para o cancro do pulmão.
Este trabalho teve como objectivo verificar esta possível associação em 175 doentes com cancro do pulmão e 217 controlos da Região Centro de Portugal, por RFLP (polimorfismo de comprimento de fragmentos de restrição).
Foi encontrada a seguinte distribuição para as frequências alélicas: 0.12 e 1.14, para os alelos mutados C e G, respectivamente, na população controlo. Os resultados não revelaram significado estatístico quando comparados com a distribuição encontrada na população de doentes. Relativamente à distribuição genotípica, a situação foi semelhante, não se registando significado estatístico, mesmo quando foram considerados genótipos de alto risco. Tal como noutras populações de diferente origem étnica, parece existir desequilíbrio de ligação para ambos os polimorfimos na população-controlo. Concluímos que nesta amostra de população portuguesa os polimorfismos m1 e m2 de CYP1A1 são particularmente raros, parecendo não existir relevância clínica nem associação à susceptibilidade ao cancro do pulmão.
Palavras-chave: Cancro do pulmão, tabaco, citocromo p450, CYP1A1, desequilíbrio de ligação.
Introduction
Cancer arises as a result of a complex sequence of mutational events that contributes to the breakdown of regulatory mechanisms, such as cell cycle control, apoptosis and differentiation. Some of the genes involved in these mechanisms, such as TP53, KRAS, or RB1, may become mutated after environmental carcinogenic exposure. Inter-individual variations in the activity of detoxifying enzymes such as cytochrome P450 enzymes, glutathione S-transferase (GSTs) and N-acetyltransferases (NATs) are also important in susceptibility to cancer1-4. Genetic polymorphisms explain these functional differences.
The search for susceptible genetic profiles is a new and promising field of preventive medicine. Two main approaches have been used: the search for specific candidate genes and genome-wide searches based on microarray technologies. We followed the former, classical approach to search for genetic polymorphisms associated with susceptibility to sporadic lung cancer.
Lung cancer is the leading worldwide cause of mortality5,6. In Portugal it is the third highest cause of death and the leading cause of cancer death. In lung cancer, both exposure to chemical agents, such as polycyclic aromatic hydrocarbons (PAH) in cigarette smoke and nitrosamines and inherited differences in metabolic capacity are thought to play a primary role in carcinogenesis7,8.
Smoking appears to be responsible for 85% of lung cancer in the Portuguese population9. In addition to tobacco compounds, other carcinogens such as alcohol, radon, silica and diesel exhaust particles may also contribute10,11.
The cytochrome P450 is a multi-enzymatic system involved in Phase I metabolism of a wide range of structurally diverse substrates12.
P450 enzymes insert molecular oxygen, increasing the hydrophility and excretion of substrates from the cell13. The enzyme aryl hydrocarbon hydroxilase (AHH), codified by the CYP1A1 gene, belongs to the cytochrome P450 system and is responsible for the activation of PAH such as benzo(a)pyrene and coronene14. More specifically, AHH catalyses the transformation of PAH to the BP-7.8-epoxide form which is further oxidised to BP-7.8-dihydrodiol by the epoxide hydrolyse (EH) enzyme. BP-7.8-dihydrodiol is then transformed by AHH to BP-7.8-dihydrodiol-9.10-epoxide15,16.
PAH metabolites are powerful carcinogens responsible for countless mutations present in squamous cell carcinomas (SCC) of the lung. PAH also work as powerful inducers of CYP1A1 gene product by binding to a citosolic receptor, the transcription factor aryl hydrocarbon receptor that ultimately activates and regulates the expression of the gene13,17-19.
It has been demonstrated that AHH expression is induced in the bronchial airways of more than 80% of lung cancer patients who are smokers20, presumably due to the high concentration of PAH and other carcinogens present in tobacco smoke. As a result, the relative levels of CYP1A1 expression in bronchi have been linked to the pathogenesis of tobacco induced lung cancer.
CYP1A1 gene is located in chromosome 15q22–q24 and in addition to the lung it is also expressed in the liver, gastrointestinal tract, brain, lymphocytes and macrophages21,22.
Four different polymorphisms of CYP1A1 gene, m1, m2, m3 and m4 have been described23 and the m4 polymorphism has a very low frequency in Caucasian populations.
The m1 and m2 polymorphisms are more widely studied not only due to their higher genotype frequency but also their possible involvement in lung carcinogenesis24.
The m1 polymorphism, involving an MspI restriction site, is a T6235C transition in the 3’ non-coding region of the gene, 250 bp downstream from the polyadenylation site20. The nucleotide substitution appears to be associated with increasing levels of CYP1A1 expression in response to PAH25.
The m2 polymorphism is an A4889G transition in exon 7 leading to an isoleucine to a valine exchange and involves a BsrDI restriction site26. The amino acid exchange lies within the hem-binding region of the protein and has been shown to increase the catalytic activity of the enzyme seven-fold27.
As AHH activates procarcino genes, individuals with high CYP1A1 gene inducibility or increased enzymatic activity may be more susceptible to the carcinogens of tobacco smoke and to developing lung cancer. Recent studies have suggested that m2 and m1 polymorphisms are probably involved in linkage disequilibrium25,28, but this remains a controversial issue.
The present study aimed to evaluate the importance of CYP1A1 polymorphisms as susceptibility factors for lung cancer and to verify the existence of the allelic linkage disequilibrium.
Materials and methods
Cases and controls
The lung cancer cases consisted of 175 patients (130 males and 45 females; average age ± SD = 62.1 ± 10.2 yrs) who were followed in the Pulmonology Department of the Hospital da Universidade de Coimbra, Portugal.
Histological diagnosis was performed according to the WHO 2004 classification. The control group included 217 unrelated volunteers (143 males and 74 females; mean age ± SD = 70.8 ± 11.8 yrs) who had no history of cancer and as age and gender have never been shown to influence CYP 450 polymorphisms29 .
All individuals were from Portugal’s midlands region and Caucasian. Informed consent from patients, as well as from controls was obtained together with permission from the Ethics Committee.
Methods
DNA was extracted from frozen peripheral blood using the urea DNA extraction technique.
DNA concentration was verified using a spectrophotometer (UV-160A-Shimadzu). CYP1A1 m1 and m2 genotypes were characterised using RFLP assay.
For m1 polymorphism, a PCR was carried out in a total volume of 50μl containing 200 ng of DNA, 1x reaction buffer, 1.5 mM of MgCl2 , 200 μM of dNTPs, 5 % of dimethyl sulfoxide, 0.2 μM of primer forward (5’ TAG GAG TCT TGT CTC ATG CCT 3’), 0.2 μM of primer reverse (5’ CAG TGA AGA GGT GTA GCC GCT 3’), and 1U Taq DNA Polymerase (Bioline). The samples were amplified using a thermal cycler with na initial denaturation at 94ºC for 5 mins followed by 30 cycles with denaturation at 94ºC for 30 seconds, annealing at 62ºC for 60 seconds and primer extension at 72ºC for 60 seconds, followed by a final extension step at 72ºC for 5 mins. The PCR products were then analysed in a 1% agarose gel with ethidium bromide and visualised using a UV transilluminator.
For RFLP assay, digestion was carried out overnight at 37ºC, in a total volume of 15 μl containing 5 μl of PCR product, 1x buffer and 3U Msp I In the pre sence of the 6235C polymorphism, the enzyme MspI, digested the 340 bp PCR pro duct in two bands of 200 and 140 bp. The wild type 6235C form corresponds to the absence of the restriction site.
For CYP1A1 m2, the PCR was carried out in a total volume 50 μl containing 200 ng of DNA, 1x reaction buffer, 1.5mM of MgCl2, 200 μM of dNTPs, 5% of dimethyl sulfoxide, 0.2 μM of primer forward 5’ CAG ACC AGG TAG ACA GAG 3’, 0,2 μM of primer reverse 5’ GTC CAC CCT CTT AAG CTC T 3’ and 1U Taq DNA Polymerase (Bioline). The PCR protocol was carried out in the same thermal cycler with an initial denaturation at 94ºC for 5 mins followed by 30 cycles of denaturation at 94ºC for 30 seconds, annealing at 61ºC for 60 seconds and primer extension at 72ºC for 60 seconds. The PCR products were analysed in a 1% agarose gel with ethidium bromide and visualised using a UV transilluminator.
For RFLP assay, digestion was carried out overnight at 65ºC in a total volume of 15 μl containing 5 μl of PCR product, 1x buffer and 4U of BsrDI. In the presence of the 4889G polymorphism, the enzyme BsrDI, digested the 350 bp PCR product in two bands of 232 and 128 bp.
The 4889A wild type polymorphism corresponds to the absence of the restriction site.
Statistical analysis
The odds ratio (OR) with the corresponding 95% confidence interval (CI) and Pearson chi-square test of independence (c2) were used in data analysis. The level of significance was set at p<0.05.
Results
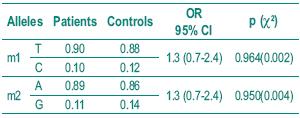
The histological diagnosis included squamous cell carcinoma, adenocarcinoma and small cell carcinoma. Table I shows the frequency of the histological types studied. Table II depicts the allelic frequencies in the tumoral and control populations. There were no statistically significant differences in the allelic frequencies in the tumoral and control group.
Table I – Distribution of histological diagnosis in patients
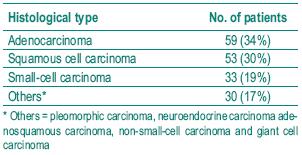
Table II – Distribution of allelic frequencies in the two groups
The distribution of genotype frequencies for both polymorphisms is shown in Table III.
Table III – Distribution of genotype frequencies
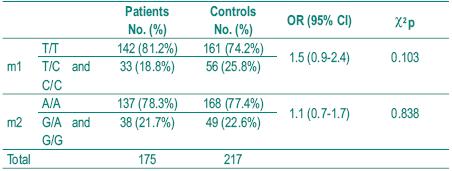
As it is rare to find patients homozygous for the variant alleles C and G, homozygous and heterozygous were analysed together. In the patient group there were only 3 individuals homozygous for C allele and 2 homozygous for G allele and in the control group there were only 4 homozygous for C allele and 1 for G allele. There were also no statistically significant differences between the two groups. That were shown to be in the Hardy-Weinberg equilibrium (p>0.05). We also analysed the frequency of individuals simultaneously homozygous for both polymorphisms associated with lower enzymatic activity (TT and GG), expecting they would be represented higher in the control group, but no significant difference was found between patients and controls. Fig. 1 illustrates the analysis of genotypes using RFLP assays.
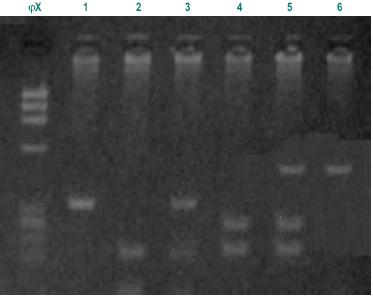
Fig. 1 – Different fragments from Msp I and Bsr DI digestions. ?X molecular weight; 1-m1:TT, homozygous wild type (340bp); 2-m1: homozygous CC (200bp and 140bp fragments); 3-m1: heterozygous TC (340bp, 200bp and140bp fragments); 4-m2: AA, homozygous wild type (350bp fragment); 5-m2: heterozygous AG, (350bp, 232bp and 118bp fragments); 6–m2: GG, homozygous double mutated (232bp,118bp fragments)
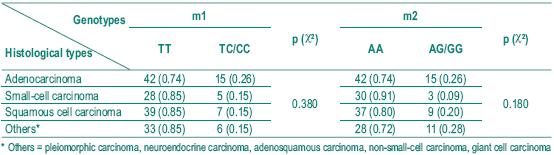
Among the different histological types of lung cancer, as squamous cell carcinoma is supposed to be more frequently associated with environmental toxic exposure, we analysed any difference between the distribution of the genotypes in the different types of carcinomas (Table IV). No statistically significant difference was found. Nevertheless, we found a small statistical significance (p=0.049) when we compared the allelic distribution for m2 allele in adenocarcinoma and small-cell carcinoma, and when we considered the small and squamous cell association and the other histological types (p=0.058).
Table IV – Distribution of genotypes in different histological types of lung cancer
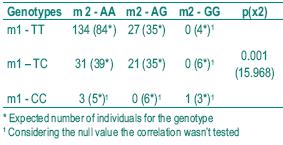
Although our samples included very few homozygous genotypes, we analysed if there was any suggestion of allele linkage disequilibrium in the control population for the higher risk genotypes. The results shown in Table V, where the number of observed and expected individuals is compared for each combined genotype, support the existence of linkage disequilibrium.
Table V – Linkage disequilibrium analysis in the control population
Discussion
The aim of this work was to define a correlation between the two most widely studied CYP1A1 gene polymorphisms, m1 allele (T6235C transition) and m2 allele (A4889G transition) and susceptibility to lung cancer.
The allele and genotype frequencies found in our population samples were quite similar to those described in other Caucasian populations30-33. The low frequency of the high-risk genotypes in Caucasian populations complicates data analysis. Only in populations of Asian origin do the frequencies of C and G alleles and of correspondent homozygous genotypes have higher frequencies.
For example, frequency for the m1 heterozygous is 0.171 in our lung cancer population is half of the frequency found in the Chinese population (0.395). We also describe a frequency of 0.017 for m1 homozygous, while in the Mongol population the frequency is 0.22817.
The results described in Table III analyse the association between the two polymorphisms m1 and m2 and the risk of lung cancer.
Though no statistically significant difference could be found (p>0.05), the unexpected fact is that the homozygous genotype for the wild genotypes was more frequent in patients, especially for m1 polymorphism (OR=1.5; 95% CI 0.9-2.4). It is likely that results would be different with more extended samples including more homozygous for the mutant high-risk genotypes.
We also found no association between the major histological types (Table IV).
Nevertheless when we analysed the allelic distribution of m2 polymorphism, we found a small statistical significance in terms of adenocarcinoma and small-cell carcinoma (p=0.049) and also between small and squamous cells and the other histological types (p=0.058).
In Asian populations, where the mutant alleles reach higher frequencies, CYP1A1 m1 and m2 polymorphisms have been widely described as genetic susceptibility factors for environmentally associated lung cancer for both squamous cell and adenocarcinoma20,34,35.
An association between the presence of at least one copy of the mutant m1 allele and increased risk for squamous cell carcinoma (OR=2.4; 95% CI of 1.2-4.7) was found in more mixed populations, including Japanese, Hawaiian and Caucasians by Le Marchand et al.36. No statistically significant difference was found for the mutant m2 allele in terms of lung cancer in general and for other histological types. In Caucasian populations, results remain controversial and many other authors also could not find any association30,32,33. Other investigators studied both CYP1A1 polymorphism and Phase II enzyme polymorphisms, known to influence excretion of genotoxic compounds. Montserrat Garcia-Closas et al.31 found that while neither the CYP1A1 MspI heterozygous genotype alone nor the GSTM1 null genotype alone were associated with a significant increase in lung cancer risk, both genetic traits are associated with a twofold increase in risk (95% CI 1.0-3.4).
The inconsistency of results remains when the association between genotypes, the dose level of tobacco exposure and susceptibility to lung cancer is studied. For instance, Nakachi et al.37 showed that individuals with the m1 susceptible genotype were at a remarkably higher risk at a low dose level of cigarette smoke (OR=7.31; 95% CI 2.13-25.12) and that the difference in susceptibility between genotypes was reduced at high dose levels. On the contrary, Montserrat Garcia-Closas et al.31 could not find enough evidence for a substantial modification of the effect of tobacco pack-years on lung cancer risk by the CYP1A1 m1 and GSTM1 genotypes. In a pooled analysis, Le Marchand et al.27 found a trend to an overrepresentation of m1 polymorphism in lung cancer patients, especially in non-smokers and in women.
In multifactorial cancer, the risk conferred by these polymorphisms, generally called SNPs (from single nucleotide polymorphisms), is typically small to moderate, with small odds ratios generally with wide confidence intervals. Results are also often conflicting.
One cause for these discrepancies is the sample size. As these are low penetrance polymorphisms, each one having little influence on the overall risk, large population studies are needed. But even within populations of thousands of individuals, results often remain inconclusive. Recently, microarray technology has allowed a simultaneous study of multiple polymorphisms and hopefully it will allow the discovery of genetic profiles for disease predisposition38, even though these technologies are very expensive, demand huge population samples and are very prone to statistical errors.
The possibility of allelic linkage disequilibrium for m1 and m2 genotypes has been suggested but never confirmed. In our control sample we also found a suggestion of the linkage (p< 0.001), explaining the similarity of results for the two alleles. Linkage disequilibrium has been proved to be associated with evolutionary mechanisms that select functionally important loci39.
In the future some other important issues must be studied. An important point is the relation between smoking and CYP1A1 polymorphisms. The time of exposure, the number of cigarettes and the age at starting smoking are important to confirm the relation between the polymorphisms and the increasing susceptibility to developing lung cancer. These issues are still under debate and controversial results have been published.
The relationship between Phase I enzymes such as CYP1A1 and Phase II enzymes such as GSTM1 is another important parameter for understanding the influence of the activation and detoxification of carcinogens and the development of lung cancer.
The study of other regulator genes such as GSTM1, CYP2D6 and NAT2 and their association with the presence of mutations in the tumour suppressor gene TP53 is already in course and will allow a better understanding of lung cancer carcinogenesis.
Future studies should also include environmental exposure to other toxins, as well as diet factors such as alcohol.
In conclusion, we did not find any statistically significant difference in allelic and geno type frequency in the populations studied.
Apparently there is no association between CYP1A1 m1 and m2 polymorphisms and susceptibility to lung cancer. Nevertheless, the p values found between squamous and small-cell and the other histological types and also for adenocarcinoma and small-cell confirm the controversial results involving these studies and suggest that the population should be extended to obtain unequivocal results. As the statistical analysis suggests the existence of allele linkage disequilibrium in the Portuguese population, the simultaneous study of the two alleles in future investigations seem unnecessary.
Acknowledgements
We are grateful to Prof. Luís Almeida and Dr. Luís Mesquita for their support and expertise and to Filipa Carvalhal Marques, Patrícia Couceiro, Pedro Pinto and Tatiana Coelho for their amazing support and work.
Bibliography
1. Hartwell LH, Kastan MB. Cell cycle control and cancer. Science 1994; 266:1821-1828.
2. Kastan MB, Canman CE, Leonard CJ. P53, cell cycle control and apoptosis: implications for cancer. Cancer Metastasis Rev 1995; 14:3-15.
3. Kinzler KW, Vagelstein B. Gatekeepers and caretakers. Nature 1997; 386:761-763.
4. Mukhopadhyay T, Maxwell SA, Roth JA. P53 suppressor gene. Austin (TX): R. G. Landes Co, 1995.
5. Stewart BW, Kleihues P. World cancer report. Lyon: IARC Press, 2003; 182.
6. Ng DPK, Tan KW, Zhao B, et al. CYP1A1 polymorphisms and risk of lung cancer in non-smoking Chinese women: influence of environmental tobacco smoke exposure and GSTM1/T1 genetic variation. Cancer Causes and Control 2005; 16:399-405.
7. Lee KM, Kang Daehee, Clapper ML et al. CYP1A1, GSTM1, and GSTT1 polymorphisms, smoking, and lung cancer risk in a pooled analysis among asian populations. Cancer Epidemiol Biomarkers Prev 2008; 17(5):100-125.
8. Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet 2007; 32:425.431.
9. Parente B, Queiroga H, Teixeira E, et al. Epidemiological study of lung cancer in Portugal (2000/2002). Rev Port Pneumol 2007; 13(2):255-265. [ Links ]
10. Takano H, Yanagisawa R, Ichinose T, et al. Lung expression of cytochrome P450 1A1 as a possible biomarker of exposure to diesel exhaust particles. Arch Toxicol, 2002; 76:146-151.
11. McClellan RO. Health effects of exposure to diesel exhaust particles. Ann Rev Pharmacol Toxicol 1987; 27:279-300.
12. Hirnoven A. Polymorphisms of xenobiotic-metabolizing enzymes and susceptibility to cancer. Environ Health Perspect 1999; 107:37-47.
13. Belogubova EV, Ulibina YM, Suvorova IK, et al. Combined CYP1A1/GSTM1 at-risk genotypes are overrepresented in squamous cell lung carcinoma patients but underrepresented in elderly tumor-free subjects. J Cancer Res Clin Oncol 2006; 132:327-331.
14. Gonzalez FJ, Nebert DW. Evolution of the P450 superfamily: animal plant “warfare’’, molecular drive and human genetic differences in drug oxidation. Trends in Genetics 1990; 6:182-186.
15. Kawajiri K. CYP1A1. IARC Sci Publ 1999; 148:159-72.
16. Smith G, Stubbins MJ, Harries LW, et al. Molecular genetics of the human cytochrome P450 monooxygenase superfamily. Xenobiotica 1999; 28 (12):1129-1165.
17 Korytina GF, Akhmadishina LZ, Kochetova OV, et al. Association of polymorphisms of the CYP1A1 and CYP1A2 cytochrome P450 genes with chronic obstructive pulmonary disease in Bashkortostan. Molecular Biology 2008; (42)1:27-36.
18. Spink DC, Spink BC, Cao JQ, et al. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis 1998; 19:291-298.
19. Lyakhovich VV, Vavilin VA, Zenkov NK. Menshchikova EB. Activated oxygen metabolites in monooxy-genase reactions. Bull Sib Otd Ross Akad Med Nauk 2005; 118:7-12.
20. Kawajiri K, Nakachi K, Imai K, et al. Identification of genetically high risk individuals to lung cancer by DNA polymorphisms of the cytochrome P450IA1 gene. FEBS Lett 1990; 263:131-133.
21. Hildebrand CE, Gonzalez FJ, McBride OW, et al. Assignment of the human 2,3,7,8-tetrachlorodibenzo–p dioxininducible cytochrome P1 -450 gene to chromosome 15. Nucleic Acids Res 1985; 13: 2009-2016.
22. Song BJ. Friedman FK. Park SS, et al. Monoclonal anti body -directed radioimmunoassay detects cytochrome P -450 in human placenta and lymphocytes. Scien ce 1985; 228: 490-492.
23. Crofts F, Cosma GN, Currie D, et al. A novel CYP1A1 gene polymorphism in African – Americans. Carcinogenesis 1993; 14:1729-1731.
24. Masson LF, Sharp L, Cotton SC, et al. Cytochrome P-450 1A1 Gene Polymorphisms and risk of breast cancer: A HuGE review. American Jornal of Epidemiology 2005; 161(10):245-263.
25. Landi MT, Bertazzi PA, Shields PG, et al. Association between CYP1A1 genotype, mRNA expression and enzymatic activity in Humans. Pharmacogenetics 1994; 4:242-246.
26. McGrath M, Hankinson SE. Cytochrome P450 1A1, cigarette smoking, and risk of endometrial cancer. Cancer Causes Control 2007; 18:1123-1130.
27. Le Marchand L, Guo C, Benhamou S, et al. Pooled analysis of the CYP1A1 exon 7 polymorphism and lung cancer. Cancer Causes and Control 2003; 14:339-346.
28. Nerurkar PV, Okinaka L, Aoki C, et al. CYP1A1, and GSTP1 genetic polymorphisms and urinary 1 -hydroxypyrene excretion in non -occupationally exposed individuals. Cancer Epidemiol Biomarkers Prev 2000; 9:1119-1122.
29. Garte S, Gaspari L, Alexandrie AK, et al. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev 2001; 10:1239-1248.
30. Drakoulis N, Cascorbi I, Brokmoller J, et al. Polymorphisms in the human CYP1A1 gene as susceptibility factors for lung cancer: exon -7 mutation (4889 A to G), and a T to C mutation in the 3’-flanking region. Clin Investig 1994; 72:240-248.
31. Garcia-Closas M, Kelsey KT, Wiencke JK, et al. A case-control study of cytochrome P450 1A1, glutathione S -transferase M1, cigarette smoking and lung cancer susceptibility. Cancer Cases and Control 1997; 8:544-553.
32. Shields PG, Caporoso NE, Falk RT, et al. Lung cancer, race and a CYP1A1 genetic polymorphism. Cancer Epidemiol Biomark Prev 1993; 2:48-485.
33. Sugimura H, Hamada GS, Suzuki I, et al. CYP1A1 and CYP2E1 polymorphisms and lung cancer, casecontrol study in Rio de Janeiro, Brazil. Pharmacogenetics 1995; 5:S145-S148.
34. Hayashi SI, Watanable J, Nakachi K, Kawajiri K. Genetic linkage of lung cancer-associated MspI polymorphism with amino acid replacement in the heme binding region of the human cytochrome P4501A1 gene. J Biochem 1991; 110:407-411.
35. Nakachi K, Hayashi S, Kawajiri K, Imai K. Association of cigarette smoking and CYP1A1 polymorphisms with adenocarcinoma of the lung by grades of differentiation. Carcinogenesis 1995; 16:2209-2213.
36. Le Marchand L, Sivaraman L, Pierce L, et al. Associations of CYP1A1, GSTM1, CYP2E1 polymorphisms with lung cancer suggest cell type specificities to tobacco carcinogens. Cancer Researcher 1998; 58:4858-4863.
37. Nakachi K, Imai K, Hayashi S, et al. Genetic susceptibility to squamous cell carcinoma of the lung in relation to cigarette smoking dose. Cancer Res 1991; 51:5177-5180.
38. Peter I, Bakker P, Maller J, Yelensky, et al. Evaluating and improving power in whole-gene association studies using fixed marker sets. Nature Genetics 2006: 38(6):663-667.
39. Kato M, Sekine A, Ohnishi Y, et al. Linkage disequilibrium of evolutionary conserved regions in the human genome. Bio Med Central Genomics 2006; 7:326-331.
Paula Mota
Instituto de Genética.
Faculdade de Medicina
Rua Larga
3000 Coimbra
e-mail: lcarvalho@huc.min-saude.pt
Recebido para publicação/received for publication: 09.04.28
Aceite para publicação/accepted for publication: 09.06.08
