










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Pneumologia
versão impressa ISSN 0873-2159
Rev Port Pneumol v.16 n.1 Lisboa jan. 2010
Linfangioleiomiomatose – A propósito de três casos clínicos
Carla Valente¹, Sónia André¹, Alexandra Catarino², Fátima Fradinho², Fernanda Gamboa², Mário Loureiro³, M Fontes Baganha4
1 Interna Complementar de Pneumologia/Resident, Pulmonology
2 Assistente Hospitalar de Pneumologia/Consultant, Pulmonology
3 Director do Serviço de Pneumologia dos HUC/Head, Pulomonology Unit, HUC
4 Director do Departamento de Ciências Pneumológicas e Alergológicas dos HUC/Director, Dept. of Pulmonology and Allergological Sciences, HUC
Resumo
A linfangioleiomiomatose (LAM) é uma doença rara, de etiologia desconhecida, caracterizada pela proliferação anormal de células musculares lisas nas regiões perilinfática, perivascular e peribrônquica.
A LAM pode ocorrer esporadicamente ou associada ao complexo esclerose tuberosa (CET) e hamartose hereditária multiorgânica1.
Em ambas as situações a LAM afecta principalmente mulheres jovens em idade fértil, sendo que aproximadamente 1/3 das mulheres com CET têm LAM2.
A propósito desta patologia, os autores elaboram uma revisão da literatura e descrevem os casos clínicos de três doentes do sexo feminino com o diagnóstico de LAM com base nos achados clínicos e imagiológicos.
Palavras-chave: Linfangioleiomiomatose, complexo esclerose tuberosa.
Lymphangioleiomyomatosis – report of three cases
Abstract
Pulmonary lymphangioleiomyomatosis (LAM) is a rare disease of unknown aetiology. It is characterized by proliferation of abnormal smooth-muscle cells throughout the peribronchial, perivascular and perilymphatic regions of the lung.
LAM may occur sporadically, in association with tuberous sclerosis complex (TSC) or inheritable multiorgan hamartomatosis 1.
In either situation, LAM occurs almost exclusively in women of reproductive age, and approximately one third of the patients with TSC have LAM2. The authors review the cases of three female patients diagnosed with LAM based on clinical and radiological findings. A brief review of the disease is then presented.
Key-words: Lymphangioleiomyomatosis, tuberous sclero sis complex.
Introdução
A linfangioleiomiomatose ou linfangiomiomatose (LAM) é uma doença rara que afecta principalmente mulheres na idade reprodutiva.
Caracteriza-se pela proliferação de células com fenótipo de músculo liso (células LAM), habitualmente observadas nas áreas peribrônquicas, perivasculares e perilinfáticas. A proliferação de células LAM pode obstruir os bronquíolos, conduzindo a possível obstrução aérea, air trapping, formação de bolhas, lesões quísticas e pneumotóraces. A obstrução dos vasos linfáticos pode resultar em quilotórax e ascite quilosa e a obstrução das vénulas pode levar a hemossiderose e hemoptises.
Nesta patologia verifica-se também uma excessiva actividade proteolítica, responsável pela formação de lesões quísticas e destruição pulmonar.
Em 1966, Corong e Enterline foram os primeiros a descrever detalhadamente um grupo de 20 doentes com diagnóstico de LAM3,4.
A LAM é uma entidade clínica distinta da linfangiomatose pulmonar difusa, em que se verifica proliferação dos vasos linfáticos de parede fina, que se encontram ectasiados, irregulares e com células musculares lisas na sua parede.
Devido à raridade da doença e à sua inespecificidade clínica, a LAM pulmonar pode ser de forma errónea confundida com asma, doença pulmonar obstrutiva crónica ou doenças intersticiais crónicas inespecíficas, podendo por esse motivo verificar-se um atraso no diagnóstico de cerca de cinco anos após a primeira apresentação clínica, pelo que esta doença deve ser considerada em todas as mulheres em idade fértil que apresentem sintomas pulmonares.
A LAM ocorre em duas formas: a esporádica (mais comum) e a associada ao complexo esclerose tuberosa (CET), doença autossómica dominante, caracterizada pelo crescimento de hamartomas cerebrais, renais, cutâneos, cardíacos e pulmonares, resultando de mutações da linha germinativa de ambos os genes TSC1 e TSC2 do cromossoma 9q34 e 16p13, respectivamente6.
As células presentes na LAM pulmonar são do tipo músculo liso, positivas para os seus marcadores tecidulares e não são malignas3,4.
Estas células apresentam a particularidade de reagirem com o anticorpo monoclonal HMB45; contudo, existe uma significativa variabilidade quanto à sua positividade e esta não é absolutamente necessária para o diagnóstico de LAM7.
A proliferação de músculo liso em redor dos brônquios origina a diminuição do seu lúmen e consequente obstrução do fluxo aéreo, em parte responsável pelo aparecimento de quistos pulmonares, tendo Sobonya et al 8 demonstrado que a perda de matriz extracelular a nível alveolar é a explicação para o colapso e irregularidade das vias aéreas circundantes.
O facto de quase todos os doentes com LAM associada ou não à esclerose tuberosa serem mulheres9, tendo sido documentada a exacerbação da doença após administração de estrogénios10, levou à implicação destes na sua patogénese, pelo que é de evitar o uso de contraceptivos orais.
Tem-se verificado um aumento do número de doentes diagnosticadas após a menopausa, sendo que quase todas estão sob terapêutica hormonal de substituição com estrogénios12.
A idade do diagnóstico é variável, sendo mais habitual entre a puberdade e a menopausa, no entanto, na literatura encontra-se referência a uma jovem com 11 anos e a uma mulher com 769. Não parece haver relação causal entre tabagismo e LAM, embora nas mulheres fumadoras se possa constatar um agravamento da função pulmonar preexistente.
A gravidez pode exacerbar a doença, sendo que globalmente aumenta em 11 vezes o número de complicações13, pelo que a gravidez é desaconselhada.
A doença afecta sobretudo o pulmão, contudo outros órgãos, como rins, gânglios linfáticos retroperitoneais, fígado, útero e pâncreas são frequentemente envolvidos5.
As manifestações pulmonares são a forma de apresentação em mais de 90% dos casos, sendo a dispneia de carácter progressivo o sintoma mais comum. A astenia é um sintoma frequente na LAM, ocorrendo em cerca de 72% dos doentes, sendo o sintoma mais comum nos idosos com esclerose tuberosa associada12. Tosse e pieira são também manifestações frequentes.
O pneumotórax é muito frequente, podendo ter carácter recorrente. Menos habituais são quilotórax, quiloptises, ascite quilosa e derrame pericárdico.
Nos doentes com associação de esclerose tuberosa e LAM podemos identificar, com significativa prevalência, angiomiolipomas renais.
O exame físico pode não ser esclarecedor. Crepitações e roncos são detectados à auscultação pulmonar em 22% e 14% dos doentes, respectivamente. O hipocratismo digital é raro e a evidência de derrame pleural ou ascite podem ser encontrados14.
O estudo funcional ventilatório (EFV) em doentes com LAM é variável. Numa análise efectuada em 35 doentes5, 51% revelavam síndroma obstrutiva, alguns com prova de broncodilatação positiva (26%), e 17% revelavam síndroma mista e 9% síndroma restritiva.
A diminuição da capacidade de difusão do monóxido de carbono (DLCO) é a alteração mais comum (83%), seguida de hipoxemia na gasometria arterial (57%)
A hipercapnia é rara nesta patologia e geralmente desenvolve-se em estádios terminais.
Um importante achado funcional ventilatório que pode diferenciar a LAM de outras doenças pulmonares intersticiais é a presença de volumes pulmonares normais ou aumentados.
Na telerradiografia do tórax esta entidade caracteriza-se por opacidades reticulares intersticiais, que podem ser subtis ou óbvias, e podem preceder e/ou acompanhar pneumotórax ou quilotórax.
A tomografia computorizada de alta resolução (TCAR) do tórax revela lesões quísticas dispersas por todo o parênquima, sendo estas habitualmente de paredes finas e regulares, com localização peribrônquica.
O prognóstico agrava-se com a extensão das lesões quísticas no parênquima pulmonar, podendo avaliar-se o grau de gravidade, consoante a área de pulmão envolvido: Grau 1 – menos de 25% do pulmão envolvido, Grau 2 – entre 25 a 50% e Grau 3 – mais de 50% do parênquima afectado pelas lesões quísticas.
A TCAR faz o diagnóstico diferencial com granulomatose de Langerhans (histiocitose X), uma vez que esta não atinge os andares inferiores nem a região costofrénica. Em alguns casos é necessário recorrer à biópsia pulmonar para obter o diagnóstico definitivo, tal como na sarcoidose pulmonar, na síndroma de Sjoegren primária, na linfangiomatose difusa, nos linfangiomiomas ou nos hamartomas quísticos.
Apesar da ausência de comprovação quanto à eficácia, várias terapêuticas antiestrogénio têm sido tentadas, como a medroxiprogesterona, o tamoxifeno e análogos da hormona luteinizante, bem como a ooforectomia14 e a radioterapia.
A terapêutica corticosteróide e imunossupressora não parece ter efeito na sobrevida dos doentes com LAM.
O transplante pulmonar oferece resultados equivalentes ou melhores aos dos obtidos pelos doentes submetidos a este procedimento por outras indicações15, no entanto a doença pode reaparecer no pulmão transplantado, o que é apesar de tudo menos frequente do que na sarcoidose16.
A LAM tem um prognóstico reservado com evolução progressiva para insuficiência respiratória e morte, sendo variável o tempo que decorre do diagnóstico aos estádios terminais da doença.
Actualmente a sobrevida estima-se em 80% e 70%, aos 5 e 10 anos de diagnóstico de doença, respectivamente. A sobrevida pode melhorar com o diagnóstico mais precoce ou com o diagnóstico de doença mais benigna.
A propósito desta patologia, os autores descrevem os casos clínicos de três mulheres com LAM.
Caso 1
Doente do sexo feminino, 51 anos, raça caucasiana, oligofrénica, não fumadora, referenciada à consulta de Pneumologia em 2001 por toracalgia direita, dispneia a médios esforços, astenia e emagrecimento (não quantificado). Dos antecedentes pessoais, de referir a menarca aos 12 anos, sem história de gravidez. Sem antecedentes de patologia pulmonar ou cardíaca. Dos hábitos é de realçar o facto de ser não fumadora.
Medicada habitualmente com Akineton®, Bunil®, Risperdal® e Metamidol®. Ao exame objectivo a doente apresentava–se consciente, orientada, colaborante, eupneica, apirética e normotensa. À auscultação pulmonar apresentava um murmúrio vesicular globalmente diminuído, sem ruídos adventícios, e a auscultação cardíaca era rítmica e sem sopros audíveis.
Procedeu-se ao estudo complementar, do qual se destacam os seguintes exames de diagnóstico: – PCR de 0,6 mg/dl, Hb de 12,8 g/dl e leucócitos de 9200/μl;
– Gasometria arterial (FiO2 21%) com valores normais;
– EFV evidenciando uma síndroma restritiva moderada (CVF – 61,4%, VEMS – 62,0%, VEMS/CVF – 99;
– Autoanticorpos negativos; – Proteinograma electroforético, imunoglobulinas e imunocomplexos com valores normais;
– TCAR do tórax revelando imagens quísticas dispersas em ambos os pulmões compativeis com LAM. Mediastino centrado sem formações ganglionares, com caracteríticas dimensionais significativas. Ausência de derrame pleural (Fig. 1). Desconhece-se a evolução clínica, pois a doente deixou de comparecer à consulta de Pneumologia.
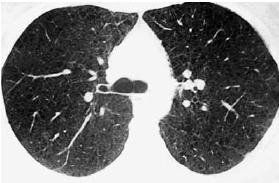
Fig. 1 – TCAR do tórax: imagens quísticas em ambos os campos pulmonares
Caso 2
Doente do sexo feminino, 42 anos, raça caucasiana, internada no Serviço de Pneumologia em 1994 com diagnóstico de quilotórax. Clinicamente referia apenas tosse seca persistente, negando dispneia ou astenia. Medicada habitualmente com Mucospas®, sem hábitos tabágicos. Sem história de alergias. História ginecológica: Menarca – 14 anos, G0P0. Nos antecedentes pessoais, de referir acidente de viação dois meses antes do internamento. Antecedentes familiares irrelevantes. Foram solicitados vários exames complementares de diagnóstico, dos quais se destacam os seguintes:
– PCR de 1,4 mg/dl, Hb de 11,7 g/dl e leucócitos de 8600/μl;
– Gasometria arterial (FiO2 21%) com valores normais;
– Bioquímica do líquido pleural com triglicéridos de 1781g/dl;
– EFV mostrando uma síndroma obstrutiva ligeira (CVF – 96,4%, VEMS – 71,0%, VEMS/CVF – 73,6, CPT – 95,1%), com prova de broncodilatação negativa;
– Autoanticorpos negativos;
– Proteinograma electroforético e imunoglobulinas com valores normais;
– TCAR do tórax com múltiplas formações quísticas de tamanho variável e parede muito fina, não confluentes, compatíveis com LAM, sem alterações pleurais ou mediastínicas significativas (Fig. 2). Colocado tubo de drenagem torácico e cumpridas medidas dietéticas, sem resolução do quilotórax, pelo que foi submetida a cirurgia para laqueação do canal torácico, tendo efectuado biópsia pulmonar no mesmo tempo cirúrgico, cuja histologia revelou parênquima pulmonar justapleural com alargamento dos espaços alveolares, com espaços quísticos grandes, alguns com espessamento do interstício devido a células musculares lisas, sem receptores de estrogénios.
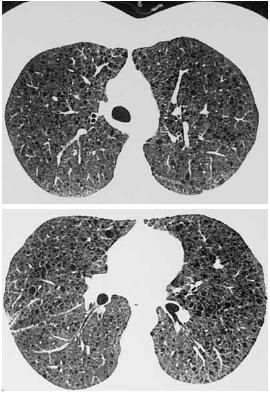
Fig. 2 – TCAR do tórax: múltiplas formações quísticas de tamanho variável, não confluentes e de parede muito fina
Pequenos nódulos periarteriolares e perivenulares. Não há lesões dos septos inter alveolares restantes nem da pleura. Alterações compatíveis com linfangioleiomiomatose (Fig. 3).
Submetida a laqueação tubar em 2002.
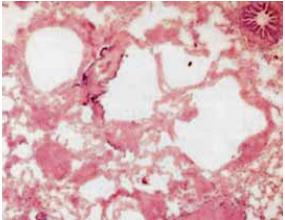
Fig. 3 – Histologia da biópsia pulmonar cirúrgica: alargamento dos espaços alveolares, com espaços quísticos grandes, alguns com espessamento do interstício devido a células musculares lisas, sem receptores de estrogénios. Pequenos nódulos periarteriolares e perivenulares. Sem lesões dos septos interalveolares restantes nem da pleura – linfangioleiomiomatose
Caso 3
Doente do sexo feminino, 34 anos, raça caucasiana, seguida em consulta de Pneumolgia desde 2003, após internamento por quadro clínico de infecção respiratória com insuficiência respiratória parcial grave.
A doente referia queixas de tosse produtiva, cansaço e dispneia progressiva para pequenos esforços com evolução desde há um ano.
Submetida, em 2002, a toracoscopia médica com biópsia pleural e talcagem por derrame pleural bilateral quiloso.
História ginecológica: Menarca – 12 anos, G1P1 (ano 2000).
Antecedentes pessoais e familiares irrelevantes. Ao exame objectivo, a doente apresentava–se consciente, orientada, colaborante, polipneica, apirética e normotensa.
À auscultação pulmonar apresentava um murmúrio vesicular globalmente diminuído, sem ruídos adventícios, e a auscultação cardíaca era rítmica e sem sopros audíveis.
Do estudo complementar, destacam-se os seguintes exames de diagnóstico:
– PCR de 0,3 mg/dl, Hb de 12,2 g/dl e leucócitos de 7700/μl;
– Gasometria arterial (FiO2 21% e em repouso): pH – 7,43; PaO2 – 52 mmHg; PaCO2 – 31 mmHg; SAT – 88%;
– EFV mostrando uma síndroma obstrutiva grave (CVF – 78,4%, VEMS – 41,1%, VEMS/CVF – 52,4, CPT – 83%);
– Autoanticorpos negativos;
– Proteinograma com valores normais;
– Imunoglobulinas e electroforese normais;
– TCAR do tórax: de forma difusa e em ambos os campos pulmonares há múltiplos quistos aéreos de várias dimensões, até 1 cm, de parede fina, sugestivos de LAM. Apresenta canal torácico ectasiado. Volumoso derrame pleural direito (Fig. 4);
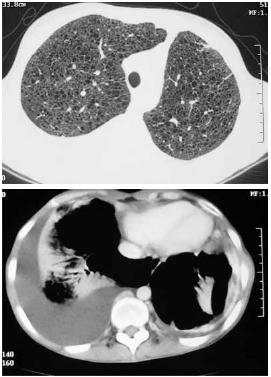
Fig. 4 – TCAR do tórax: de forma difusa e em ambos os campos pulmonares, múltiplos quistos aéreos de várias dimensões, até 1 cm, de parede fina. Canal torácico ectasiado e volumoso derrame pleural direito
– Histolologia da biópsia pleural: múltiplos fragmentos de pleura parietal mostrando colagenização, fibrina e infiltrado linfoplasmocitário com alguns PMN neutrófilos.
Evolução
A primeira doente, oligofrénica, não compareceu posteriormente às consultas de pneumologia, não sendo por este motivo possível avaliar a sua evolução clínica.
As outras duas, de 42 e 34 anos, respectivamente, encontram-se actualmente sob vigilância em consulta de pneumologia.
Ambas apresentam declínio da função ventilatória, embora sem agravamento clínico e/ou imagiológico significativo em relação à altura do diagnóstico.
A doente correspondente ao Caso clínico 2 apresenta actualmente síndroma obstrutiva moderada (CVF – 110,76%, VEMS – 64,96%, VEMS/CVF – 50,40, CPT – 113,3%, VR – 127,5%), com prova de broncodilatação negativa e DLCO de 58,6%. Está medicada com brometo de tiotrópio 18μg id e salmeterol/fluticasona 50/250 2id.
A doente correspondente ao Caso clínico 3 realizou radioterapia para castração com dose de 1200 Gy / 3 fracções que terminou em 2006, e actualmente apresenta síndroma obstrutiva grave (CVF – 72,6%, VEMS – 38,82%, VEMS/CVF – 46,17, CPT – 78,8%, VR – 116,4%), com prova de broncodilatação negativa e DLCO de 16,5%. A gasometria arterial (FiO2 21%) evidencia insuficiência respiratória parcial (pH – 7,38, PaO2 – 61,0 mmHg, PaCO2 – 39,7 mmHg, SAT – 92,7%).
Medicada com salmeterol/fluticasona 50/500 2id, está sob oxigenoterapia de longa duração no domicílio e oxigénio líquido, tendo sido colocada à doente a hipótese de transplantação pulmonar.
Discussão
A revisão destes três casos tem por objectivo lembrar o quão importante é a correlação da clínica com as alterações imagiológicas e a sua importância, não só no diagnóstico, mas também na avaliação evolutiva da linfangioleiomiomatose.
Em qualquer dos casos, a imagiologia foi fundamental para a suspeição da patologia em causa. No primeiro caso clínico descreve-se uma doente com LAM, em que o diagnóstico se baseia exclusivamente nos achados imagiológicos, bastante característicos desta patologia.
Sabe-se que as alterações na TCAR do tórax têm uma estreita correlação com a severidade da LAM e com o compromisso funcional respiratório, sendo então o parâmetro mais fiável a DLCO, o que nesta doente não foi possível avaliar, dada a sua pouca colaboração.
Salientamos a apresentação clínica no segundo caso, em que a LAM foi diagnosticada pós-ruptura traumática do canal torácico, na sequência de acidente de viação.
O quilotórax e o pneumotórax são as duas complicações major da LAM, sendo este último mais comum e diagnosticado em 39 a 53% dos doentes no início do quadro clínico e em 60 a 81% durante o curso habitual da doença17.
Em relação ao último caso clínico, a forma de apresentação não foi acidental, como no segundo caso, mas com história prévia de derrame pleural quiloso recidivante. É de referir que o início das queixas se remete a um ano após gestação, realçando o papel hormonal na progressão da doença. A ocorrência de quilotórax tem sido descrita em doentes com a forma esporádica da LAM, assim como em doentes com a associação CET e LAM. Os derrames pleurais em doentes com LAM são quase sempre quilosos1, 17.
Vários estudos sugerem que os derrames pleurais quilosos associados a LAM são mais frequentemente unilaterais do que bilaterais.
O quilotórax foi descrito em cerca de 14% dos doentes com esta patologia à data da apresentação e em 22 a 39% durante a evolução da mesma13.
Essencialmente, são três os mecanismos de formação de quilotórax: fuga de líquido quiloso (secundária a obstrução linfática proximal), fluxo transdiafragmático de ascite quilosa e passagem através dos linfáticos pleurais e vasos colaterais18.
Raramente os doentes com LAM manifestam ascite quilosa e quilúria.
Em termos de dieta, um esquema hipolipídico ou sem suplementação de triglicéridos de cadeia média tem tido pouco sucesso no controlo do quilotórax. A hiperalimentação pode diminuir a risco de fugas do líquido quiloso.
A irradiação do ducto torácico ou do mediastino não tem efeito consistente, mas a pleurodésis química tem tido sucesso na prevenção de derrames quilosos recorrentes.
Também a pleurectomia parietal tem sido eficaz nestes casos. A correcção cirúrgica do canal torácico é habitualmente eficaz na resolução do quilotórax, embora estejam descritos casos de recidiva após este procedimento.
Conclusões
A LAM é uma doença pulmonar intersticial rara, de etiologia desconhecida, que afecta quase exclusivamente mulheres, habitualmente na sua idade reprodutiva.
Geralmente o diagnóstico, que exige confirmação histopatológica, é feito entrea a 3.ª e 4.ª décadas de vida, porém os primeiros sintomas podem antecedê-lo de meses a vários anos.
Clinicamente, a sua apresentação é inespecífica, mas tosse e dispneia são os sintomas mais frequentes.
O quilotórax e o pneumotórax são as duas complicações mais frequentes da LAM. Os derrames pleurais em doentes com LAM são quase sempre quilosos e com maior frequência unilaterais.
Radiograficamente, a LAM é caracterizada por opacidades reticulares intersticiais que podem ser pouco perceptíveis ou muito exuberantes. Também podem preceder, acompanhar ou seguir um episódio de pneumotórax ou quilotórax.
A aparência da tomografia computorizada na LAM é diagnóstica quando estão presentes bilateralmente quistos, de parede fina e regular, rodeados por parênquima normal, em mulheres jovens.
Tipicamente, a alteração funcional dos doen tes com LAM consiste num padrão obstrutivo com volumes pulmonares estáticos normais ou aumentados, associado a diminuição da difusão alveolocapilar pelo CO.
O facto de afectar essencialmente mulheres em idade reprodutora sugere a existência de uma base hormonal que sustenta e favorece o aparecimento da doença, agravando-se durante a gravidez, após o parto e com o uso de terapêutica estrogénica exógena.
A possível relação da doença e o uso de estrogénios está ainda por demonstrar e estimulou, nos últimos anos, a identificação dos receptores de estrogénio e progesterona nas células de músculo liso (células LAM), cujos resultados foram contraditórios em relação à manipulação de terapêutica hormonal com o objectivo de controlar a proliferação celular e progressão da doença.
Algumas das propostas terapêuticas hormonais anti estrogénios na LAM são a ooforectomia, a irradiação do ovário, o tamoxifen (antiestrogénio), a progesterona ou a combinação de ooforectomia e progesterona ou tamoxifen e progesterona.
O prognóstico é variável, com uma sobrevida média de 70%, aos 10 anos, após o diagnóstico.
Bibliografia
1. Johnson S. Lymphangioleiomyomatosis: clinical features, management and basic mechanisms. Thorax 1999; 54:254-264.
2. Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in adult women with tuberous sclerosis complex. Mayo Clinic Proc 2000; 75:591-594.
3. Corong JL Jr, Enterline HT. Lymphangioleiomyoma, a benign lesion of chyliferous lymphantics synonymous with lymphangiopericytoma. Cancer 1966; 19:1909-1930.
4. Finlay G. The LAM cell: Am J Physiol Lung Cell Mol Physiol 2004; 286:L690-L693.
5. Chu SC, Horiba K, Usuki J, et al. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest 1999; 115:1041-1052.
6. Gomez M, Sampson J, Whittermore VH. Tuberous Sclerosis Complex. New York: Oxford University Press; 1999.
7. Hoon V, Thung SN, Kaneko M, et al. HMB reactivity in renal angiomyolipoma and lymphangioleiomyomatosis. Arch Path Lab Meth 1994; 118:732-734.
8. Sobonya RE, Quan SF, Fleishman JS, Pulmonary lymphangioleiomyomatosis: quantitative analysis of lesions producing airflow limitation. Hum Pathol 1985; 16:1122-1128.
9. Ryu JH, Moss J, Beck GJ, et al. The NHLBI lymphangioleiomyomatosis registry. Am J Resp Crit Care Med 2006; 173:105-111.
10. Yano S. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous estrogen used for infertility treatment. Thorax 2002; 57:1085 -1086.
11. Awi K, Fujikawa K, Sato T, et al. Pulmonary lymphangioleiomyomatosis. Radiat Med 1990; 8:132-135.
12. Cohen MM, Pollock-Barziv S, Johnson SR. Emerging clinical picture of lymphangioleiomyomatosis. Thorax 2005; 60:875-879.
13. Johnson SR, Tattersfield AE. Clinical experience of lymphangioleiomyomatosis in UK. Thorax 2000; 55:1052-1057.
14. Kitaichi M, Nishimura K, Itoh H, et al. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Resp Crit Care Med 1995; 151 (2, pt1):527-533.
15. Kpodonu J, Massad MG, Chaer RA, et al. The US experience with lung transplantation for pulmonary lymphangioleiomyomatosis. J Heart Lung Transplant 2005; 24:1247-1253.
16. Boehler A, Speich R, Russi EW, et al. Lung transplantation for lymphangioleiomyomatosis. N Eng J Med 1996; 335:1275-1280.
17. Sullivan EJ. Lymphangioleiomyomatosis: a review. Chest 1998; 114:1689-1703. [ Links ]
18. Corrin B, Liebow AA, Friedman PJ. Pulmonary lymphangioleiomyomatosis: a review. Am J Pathol 1975; 79:348-367.
Departamento de Ciências Pneumológicas e Alergológicas dos Hospitais da Universidade de Coimbra
Av. Bissaya Barreto e Praceta Prof. Mota Pinto
3000-075 Coimbra
e-mail: carlavalente77@yahoo.com.br
Recebido para publicação/received for publication: 09.05.04
Aceite para publicação/accepted for publication: 09.06.23
