











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Loeys-Dietz syndrome is an autosomal dominant connective tissue disease with important cardiovascular manifestations, which can lead to death at a young age due to aortic aneurysm dissection. This is a rare case of prenatal diagnosis of Loeys-Dietz syndrome, which is usually diagnosed after birth. There are three published cases on the prenatal diagnosis of this syndrome, none of them with isolated dilatation of the great vessels of the heart.
Case Report
This case reports to prenatal diagnosis in which the baby’s father suffers from Loeys Dietz syndrome with an identified pathogenic variant c.1582C > T (R528C) in TGFBR2. The father has had an aortic root replacement and has a strong family history of sudden cardiac death from acute aortic root dissection.
Given the paternal diagnosis, the couple opted to do a NIPT which confirmed that the fetus had the same TGFBR2 pathogenic variant. The couple chose to continue the pregnancy and was referred to Fetal Cardiology.
A 34-year-old primipara, had a first trimester screening with low risk and the 20-week anomaly scan was reported as normal. There were no signs of skeletal and craniofacial affection, typical manifestations of the syndrome.
At 24 weeks, the fetal echocardiography showed a dilatation of the ascending Aorta measuring 6.5 mm (z-score + 3.39) and suspected a bicuspid aortic valve, slightly echogenic leaflets with laminar flow, normal velocity. The main pulmonary artery measured 7 mm (z-score + 4.10). There was also a small pericardial effusion.
The echocardiogram performed at 29 weeks confirmed a significant disproportion between the ascending aorta, 7.6 mm (z-score +2.81) and the aortic annulus of 5.2 mm (z-score +1.64), (Figure 1). There was also a mild dilatation of the main pulmonary artery 8.5 mm (z-score +3.71) with normal velocities, no regurgitation and laminar flow (Figure 2). Although a difficult prenatal diagnosis to do, the presence of a bicuspid aortic valve was still suspected. The aortic valve leaflets were thin and there was no aortic regurgitation. There was an aneurysmal atrial septum and mild pericardial effusion.
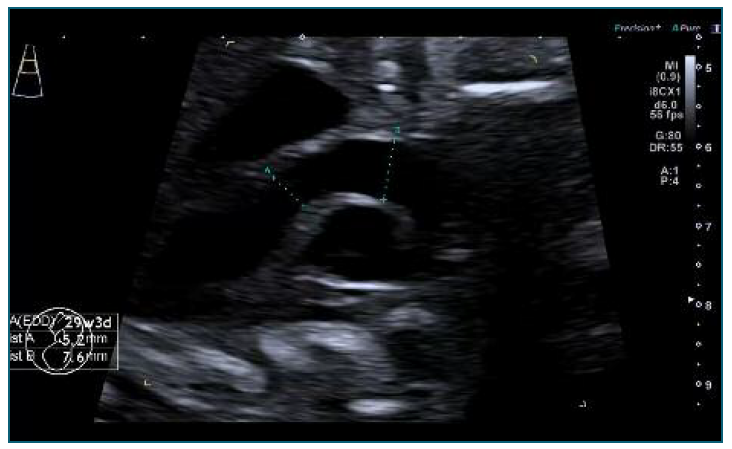
Figure 1 Transverse axial sonogram of the fetal thorax at 29 weeks: Significant disproportion between the ascending aorta, 7.6 mm (z-score +2.81) and the aortic annulus of 5.2 mm (z-score +1.64).
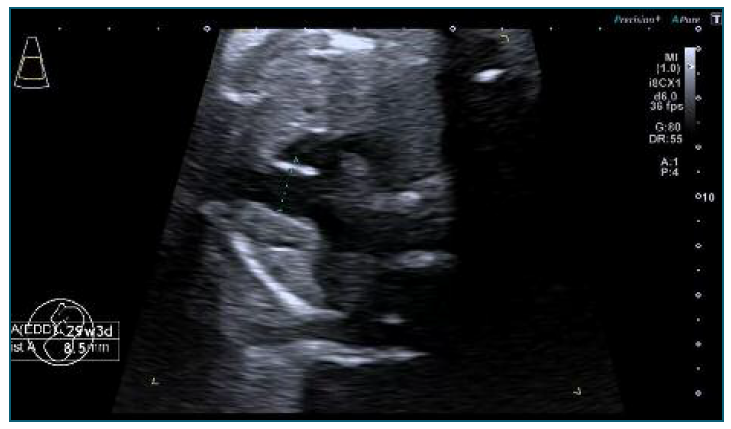
Figure 2 Transverse axial sonogram of the fetal thorax at 29 weeks: Mild dilatation of the main pulmonary artery 8.5 mm (z-score +3.71) with normal velocities, no regurgitation and laminar flow.
The z‐score were calculated using the Parameter(z) according to Krishnan et al., 2016.
The fetal Cardiology planned to refer the baby to an outpatient clinic within the 4-6 weeks of life. Mean-while, this family was counseled and referred to see a specialist in hereditary cardiomyopathies during pregnancy and explained that angiotensin receptor blockers can reduce the rate of dilation of the ascending aorta. Given the risk of recurrence in the offspring, we emphasized the importance of preconception genetic counselling, including the possibility of a preimplantation genetic test in a future pregnancy of this couple. At the last visit, at 3-4 months, the baby is asymptomatic and has normal z-scores for both main vessels. The aortic valve is tricuspid not bicuspid as suspected prenatally. The next appointment will be at one year of age.
Discussion
Loeys-Dietz syndrome is connective tissue disorder that causes aortic aneurysms and generalized arterial tortuosity, being a major cause of aortic dilatation in paediatric population and death at young age by aortic dissection1. Data concerning the prenatal diagnosis of Loeys-Dietz syndrome is scarce, but of major importance considering that aortic dissection has been reported in individuals as young as 3 months and cerebral hemorrhage as young as 3 years2. Individuals often have hypertelorism, and bifid/ broad uvula or cleft palate1.
Although it is an autosomal dominant disorder, only 25% of newly affected patients have an affected parent, while the remaining 75% have new pathogenic variants3. The diagnosis is confirmed by a molecular test, looking for pathogenic or likely pathogenic variants in one of the genes: transforming growth factor β receptor I and II (TGFBR1 and TGFBR2), decapentaplegic homolog 3 and 2 (SMAD2 and SMAD3) and the transforming growth factor β 2 and 3 ligand (TGFB2 and TGFB3) 1.
Dilatation of the great vessels of the heart is a rare finding in fetal echocardiograms. There is little knowledge about its significance, etiology and outcomes.
With regard to dilatation of ascending aorta, there are some case reports describing this condition in fetus with connective tissue diseases, being the most common of all Neonatal Marfan Syndrome4. Only three cases have been reported associated to Loeys-Dietz syndrome: s. Viassolo et al. (2006) 5, Y. Kawazu et al. (2011)6 and Gindes L et al. (2014) 7. However, the latest published cohort study about dilated ascending aorta in the fetus, showed no association between these condition and connective tissue disorders8.
Dilation of the main pulmonary artery is an even rarer condition than dilation of the ascending aorta. It has been associated with a poor prognosis in studies of diaphragmatic hernia and structural cardiac defects9, but as far as we know, this is the first time it is mentioned in the context of connective disease without structural cardiac defects.
The diagnosis of LDS was straightforward in this case due to family history, but it shows the association between connective tissue disorders and dilation of the great vessels of the heart.
In the presence of one or more of these findings: aneurysmal atrial septum, dilated main pulmonary artery and ascending aorta, referral to a fetal cardiologist and genetic counseling is recommended - these genetic conditions are serious and can be no family history of them.
Patient has given informed consent for publication of this clinical information and related pictures.
Authors’ contributions
Inês Mendes: Collected the data; Analysis the data published before; Literature review; Wrote the paper. Inês Gomes: Collected the data; Reviewing the article before submission not only for spelling and grammar but also for its intellectual content. Patrícia Caldas: Collected the data; Reviewing the article before submission not only for spelling and grammar but also for its intellectual content.

