











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Bullous congenital ichthyosiform erythroderma of Brocq or epidermolytic hyperkeratosis (EHK - MIM #113800) is a disorder of cornification, with an estimated prevalence of 1 in every 200 000 - 300 000 people1. It is a rare and severe form of congenital ichthyosis, usually with an autosomal dominant inheritance pattern2. There is no definitive or curative treatment of EHK and the therapeutic options available nowadays have limited effectiveness. In the most severe cases, it relies on the use of topical and oral retinoids3, which are teratogenic.
Case report
We report the case of a 24-year-old pregnant woman with EHK, nulliparous, single, unemployed and homeless. As reported by Morais et al. (4, at birth she presented a burned child aspect, with erythema, erosions and blisters throughout the body; she was diagnosed with EHK at 3 years of age, when she was referred to that Dermatology department. Posteriorly, at the age of 12, the genetic testing identified a variant in the gene keratin 10 (KRT10) as being responsible for her phenotype. She was followed by dermatology; however, her last appointment had been more than 3 years prior.
This patient had tried treatment with antiseptic cleansers, emollients, keratolytics, such as salicylic acid, urea or alpha-hydroxyacids, and also with the vitamin D analogue calcipotriol, with an unsatisfactory response. As a consequence, she started oral acitretin, which she took for several years. While under this treatment, the patient referred to be asymptomatic, with a completely normal skin appearance.
The patient’s pregnancy was not planned. Its diagnosis was only made at 18 weeks of gestation in the emergency department (ED) and, after that, the patient missed all the appointments scheduled, including both obstetrics and prenatal diagnosis.
She returned to the ED at 33 weeks, with complaints of abdominal pain. At that time, an ultrasound (US) was performed and revealed several fetal malformations (some of them visualized in Figure 1): abnormal facial profile with sinking of the nasal base; nasal bone unidentified; abnormal shape of the left ear with low insertion; only 4 fingers identified in the right hand (left hand was impossible to examine) and, in the genital region, an exuberant vulva. The estimated fetal weight was 1999 grams, which corresponds to the 14th percentile (of the Portuguese population). The volume of amniotic fluid and the Doppler velocimetry evaluation (of the umbilical and middle cerebral artery) were normal. She was recommended to discontinue retinoids at this time and she was referred to pediatric cardiology (for a fetal echocardiography) and, once more, to our department of prenatal diagnosis. The echocardiogram (done at 33 weeks and 3 days of gestation) was normal. The patient reported a significant clinical worsening following the suspension of acitretin. At our prenatal department, the US performed at 37 weeks and 1 day revealed a fetal growth restriction, with an estimated fetal weight of 2091 grams, 2nd percentile, with normal fetal hemodynamics evaluated by Doppler velocimetry. At this appointment, she complained of severe generalized pruritus and pelvic discomfort. On the physical examination, it was possible to see multiple scaly lesions throughout the body and hyperkeratotic plaques mostly over her abdomen and hands (Figure 2).
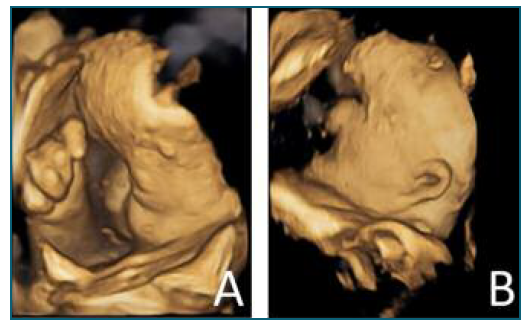
Figure 1 US (in 4D) made at 34 weeks of gestation, showing several fetal malformations: A) sinking of the nasal base; right hand with only four fingers; B) low implantation of the ears.
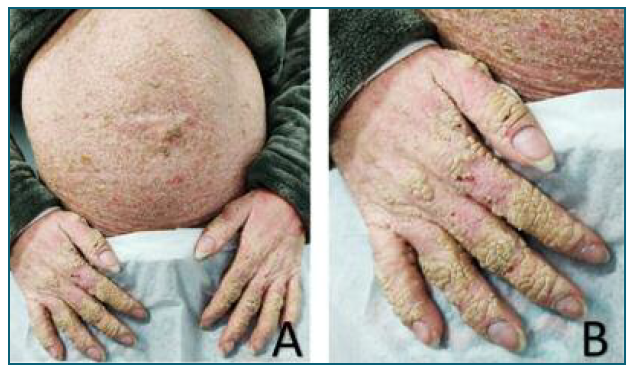
Figure 2 Clinical presentation of the patient at hospital admission. A) Hyperkeratosis more prominent in the abdomen and in her hands. B) Well-demarcated hyperkeratotic plaques with a cobblestone pattern cover the dorsal aspect of her hands.
Considering the clinical scenario, she was hospitalized at our hospital, in order to improve maternal status and carry out fetal surveillance. A multidisciplinary team guided the case with the collaboration of dermatology and anesthesia. It was decided to initiate oral antihistamines for pruritus control and topical treatment with corticoids and fusidic acid to stabilize the disease. Furthermore, the case was promptly presented to the neonatology department to predict and prepare the specific postnatal care.
Despite the fetal percentile, which would be an indication for terminating the pregnancy5, it was decided to defer the induction of labor. The objective was, at first, to optimize the maternal status: a notorious improvement of the patient’s skin was necessary to allow placement of catheters, bandages and avoid a systemic infection. The dermatology collaboration allowed a great improvement on her skin, as shown in Figure 4, with the usage of skin emollients, topical corticoid and antibiotic, in order to control bacterial infections. It was also necessary to request specific surgical fields and bandages. Her skin status also conditioned the venous accesses used. Considering her infectious risk, the best option was to use a central venous access for drugs administration, including for the anesthesia during labor.
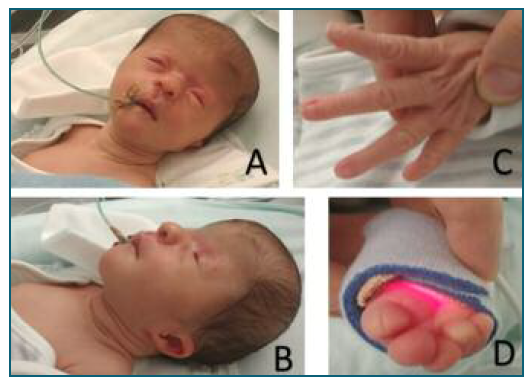
Figure 3 New born at the 2nd day of life. A, B) Craniofacial dysmorphisms: coronal craniosynostosis, with prominence of the frontal bone. Periorbital plane angioma. Microphthalmia, hypertelorism, short nose and low-set ears. C) Absence of the 1st finger of both hands. Malformed fingers (without distal phalanx). D) Changes in the toes with hallux malformation (without distal phalanx) and partial syndactyly of the 2nd and 3rd toes and of the 4th and 5th toes.
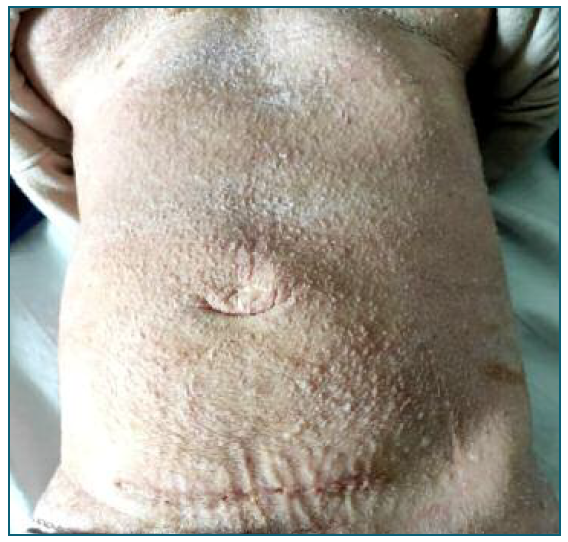
Figure 4 Patient’s abdomen after 18 days of topical treatment, showing the cesarean surgical scar. The hyperkeratosis reduced significantly and the skin became dry and scaly. The pruritus resolved and the patient was discharged with orientation to maintain cutaneous emollients.
During the hospitalization, the fetal growth and well-being were monitored, with stable sonographic parameters and reassuring cardiotocography.
At 39 weeks of gestation, it was decided to initiate the induction of labor with vaginal dinoprostone. However, due to a failed induction, two days after, a c-section was performed. She delivered a viable newborn, female, with an Apgar score of 9 at 1st and 5th minutes, weighing 2515 grams (3rd percentile).
The newborn presented multiple minor malformations, as suspected in the pre-natal ultrasound, including coronal craniosynostosis, microphthalmia, hypertelorism, short nose and low-set ears, arthrogryposis of the elbows, absence of the 1st finger of both hands and malformed fingers, equinovarus feet and genital labia majora hypertrophy, without cutaneous lesions (Figure 3). The baby was admitted to the neonatology department mainly due to feeding difficulties. After a multidisciplinary evaluation by pediatrics and genetics, it was concluded that the phenotype was compatible with retinoic acid embryopathy.
The puerpera was discharged three days after delivery, nineteen days post admission, clinically improved and with a contraceptive implant. The baby was oriented to specific medical follow up and, due to the poor socio-economic conditions, was sent to an orphanage.
Discussion
EHK is a rare dermatological disease with sparse documented experience of management during pregnancy and delivery. It is caused by a mutation in one of the genes encoding keratin (KRT1 or KRT10) (6. EHK usually manifests at birth, with redness, blistering and peeling, and can progress from a mild to a severe disease, with hyperkeratosis as the main clinical feature in adult life1,7. This patient had been diagnosed with KRT10 variant during her childhood, which is typically associated with more generalized disease8. As is evident in the presented case, it can be socially disabling, especially if there is widespread involvement of the body.
The patient had been taking acitretin for several years, after lack of improvement with different strategies. Retinoid therapy, given topically or systemically, is usually the therapeutic option for patients that show a poor response to topical drugs9. The patient mentioned that she was asymptomatic when she was under acitretin, but she abandoned her dermatological follow-up.
She did not plan the pregnancy nor did she consult any doctor when the pregnancy was diagnosed, along with missing numerous medical appointments during the pregnancy. When she was consulted by an obstetrician, at almost 34 weeks of gestation and the acitretin was suspended, there was a significant clinical worsening.
The newborn’s dysmorphic features described in this clinical case are compatible with retinoic acid embryopathy, according to the literature10,11,12.
Acitretin belongs to the retinoids class, vitamin A analogues, a class of drugs known for its teratogenic effects (FDA class X). The analogues of vitamin A, including acitretin, compete with the cellular retinoic acid receptors, interfering with the cell-cell signaling during vertebrate organogenesis. The mechanism responsible for producing many of the malformations in infants exposed to retinoic acid is an abnormality of cephalic neural crest cell activity13.
Approximately 25% of newborns submitted to retinoids in utero will develop a set of malformations, well defined as retinoic acid embryopathy12,15, which includes: cranial and facial anomalies, more often involving the ears; central nervous system defects; cardiovascular malformations and impaired development10.
These anomalies are more frequent and severe if the drug or its metabolite is present in the mother’s serum in early pregnancy, during the period of organogenesis (until about 8 weeks of gestation), which was the case. A woman undergoing treatment with acitretin should have a double contraceptive strategy and a minimum interval of 3 years is recommended between discontinuing the drug and pregnancy, which is considered to be the time necessary for the serum levels to be undetected11.
Regarding the EHK, it is important to offer the possibility of prenatal diagnosis, considering that its pattern of transmission conveys a 50% risk of having an affected child. In cases where a molecular diagnosis by genetic testing in the pregnant woman has been previously achieved, as in this case, it is possible to pursue definitive prenatal diagnosis with fetal DNA, obtained by chorionic sample biopsy or amniocentesis. In fact, EHK was the first congenital dermatosis in which the prenatal diagnosis became possible15. Moreover, it is possible to screen the fetus through fetoscopy for visual inspection along with fetal skin biopsy. In normal epidermal development, the onset of keratinization is approximately at 24 weeks. The occurrence of precocious hyperkeratosis and keratinization observed via fetal skin biopsy, before 24 weeks of gestation, can be diagnostic of epidermolytic hyperkeratosis16. In this case, only the postnatal evaluation was possible, due to the late medical evaluation. When the fetus is affected, 71% have lesions at birth7, similar to her mother’s presentation. However, the disease can be noticed later, which makes it important to establish the diagnosis even without the expected features. The baby was negative for the familiar mutation of the KRT10 gene responsible for her mother’s EHK (mutation 4724T→C, predicting the amino acid change L452P in exon 6 of KRT10 gene4). Due to the observed cranial malformations, the baby was also tested for Muenke Syndrome, and she was negative for the mutation most commonly associated with this syndrome (variant c.749C>G (p.Pro250Arg).
This patient was admitted to improve the maternal status with a multidisciplinary team, which had to take into consideration the time needed for her skin improvement, balanced with the fetal growth restriction. In consequence, in the described scenario of fetal well being assured, it was decided to delay the pregnancy termination until 39 weeks, when the mother’s skin was significantly better.
This case illustrates the importance of recognizing the teratogenic potential of drugs used by women of reproductive age, and to ensure the use of effective contraception in the ones taking this kind of medication. In addition, it is also crucial to review the medication used by pregnant women, to unsure all the teratogenic drugs are suspended on time. In the present case, the use of acicretin during most of the pregnancy conditioned a fetal polymalformative syndrome, which was, unfortunately, diagnosed late in pregnancy. Our intervention only allowed us to prevent maternal morbidity, as well as additional fetal morbidity, and provide the best follow up of the baby despite her malformations.
Authors’ contributions
Forjaz S.: Substantial contributions to the conception or design of the work; to the acquisition, and interpretation of data for the work; manuscript writing; Final approval of the version to be published. Almendra R.: contributions to the interpretation of data for the work; Review of the manuscript; Final approval of the version to be published. Braga L.: Review of the manuscript; Final approval of the version to be published. Monteiro MJ: contributions to the interpretation of data for the work; Review of the manuscript; final approval of the manuscript. Cardoso L.: contributions to the interpretation of data for the work; Review of the manuscript; final approval of the manuscript.

